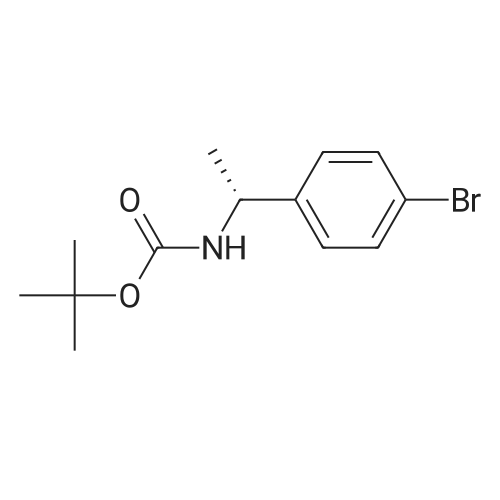
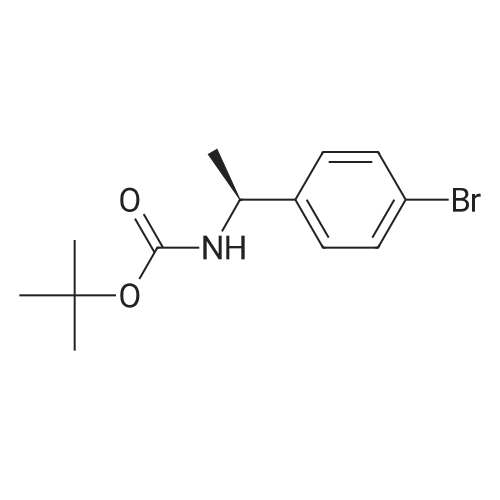
| 100% |
With triethylamine; In dichloromethane; at 0 - 25℃; |
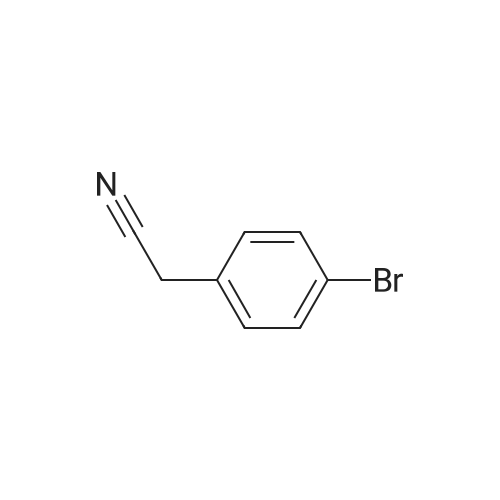
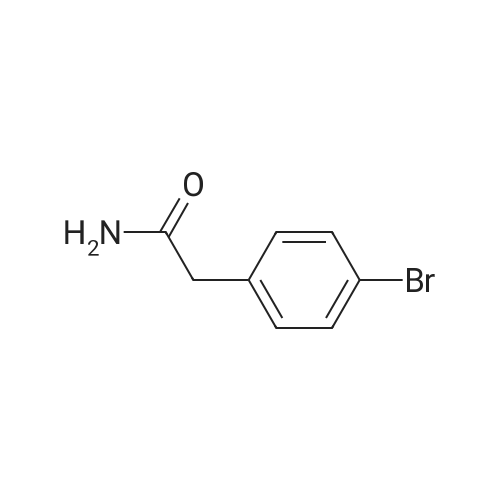
1. BOC2O (818 mg, 3.75 mmol, 1.5 eq.) was added to a stirred solution of 2-(4- bromophenyl)ethanamine (500 mg, 2.5 mmol, 1.0 eq.) and TEA (1.5 ml, 7.5 mmol, 3.0 eq.) in DCM (25 ml) at 0 0C and resulting reaction mixture is stirred at 25 0C for 6 h. The reaction mixture was diluted with DCM (75 ml), washed with water (2 x 50 ml) and brine (50 ml) and dried over Na2SO4. The solvent was evaporated under reduced pressure and the residue triturated with hexanes to yield the desired product as an off white solid.Yield: 100 % (750 mg, 2.5 mmol) |
| 98% |
In dichloromethane; at 20℃; for 3h; |
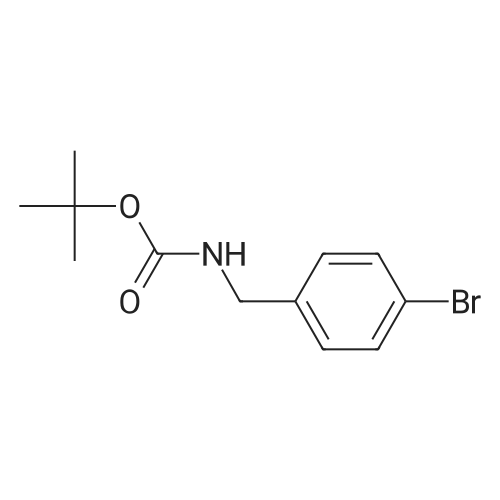
4-bromophenethylamine (10 g, 50 mm) was dissolved in 250 ml of methylene chloride, di-tert-butyl dicarbonate (12 g, 55 mmol) was added, and the mixture was stirred at room temperature for 3 hours.successively with saturated ammonium chloride solution, saturated aqueous solution of sodium bicarbonate, water, saturated salt water to wash the reaction solution, the organic phase is water-free magnesium sulfate drying reciprocation to dryness to obtain the crude product. In the title compound crystallized to obtain ethyl ether (white solid, 14.70 g, 98% yield). |
| 96.3% |
With triethylamine; In tetrahydrofuran; at 0 - 20℃; |
t°lambdatf-Butyl [2-(4-bromophenyl)ethyl]carbamate; [2-(4-Bromophenyl)ethyl] amine (2.2Og, ll.OOmmol) was dissolved in dry THF (77mL) and TEA (1.1 Ig, ll.OOmmol) was added. The mixture was cooled in an ice-bath. Di-tert-butyl dicarbonate (2.4Og, ll.OOmmol) was added. The mixture was stirred for Ih at O0C and left at rt overnight. The solvent was evaporated and column chromatography of the crude product EPO <DP n="185"/>(ISOLUTE SI (2Og), Heptane/EtOAc (90:10)) afforded the title compound (3.18g, 96.3%). 1H NMR (500MHz, CD3OD): delta 1.42 (s, 9H), 2.73 (t, 2H), 3.25 (t, 2H), 7.14 (d, 2H), 7.43 (d, 2H). |
| 95% |
With triethylamine; In dichloromethane; at 25℃; |
To a solution of 2-(4-bromophenyl)ethan-l-amine (5.00 g, 25.0 mmol) in anhydrous dichloromethane (50 mL) was added Boc20 (6.57 g, 30.1 mmol) followed by EtsN (10.4 mL, 74.9 mmol). The resulting solution was stirred overnight at 25 C and then concentrated in vacuo. The crude product was purified by FCC eluting with ethyl acetate/petroleum ether (4-bromophenethyl)carbamate as a white solid (7.1 g, 95%), LCMS (ESI, m/z): 300 [M+H]+ |
| 95% |
With triethylamine; In dichloromethane; at 25℃; |
(0935) [00330] To a solution of 2-(4-bromophenyl)ethan-l -amine (5.00 g, 25.0 mmol) in anhydrous dichlorometha e (50-raL) was added B0C2O (6,57 g, 30.1 mmol) followed by EtaN (10.4 mL, 74.9 mmol). The resulting solution was stirred overnight at 25 C and then concentrated in vacuo. The resulting crude product was purified by FCC eluting with ethyl acetate/petroleum ether (PE/EA=3 : 1) to afford fert-butyl (4-bromophenethyl)carbamate as a white solid (7.1 g, 95%). LCMS (ESI, m/z) 300 [M+H]+. |
| 91% |
In dichloromethane; at 0 - 20℃; for 3h; |
Example 3C:; Part A:; A mixture of 4-bromophenethylamine (11) (0.5 g, 2.5 mmol) and di-te/t-butyl dicarbonate (0.65 g, 3 mmol) at 00C in DCM (5 mL) was warmed to room temperature and stirred for 3 hours. LC-MS analysis indicated the reaction was complete. Oichloromethane (5 mL) was added and the solution washed with 1 lambda/ HCI (10 mL). Drying over magnesium sulfate, concentration and purification by flash column chromatography (Sitheta2, 12.5 % ethyl acetate in hexanes) afforded compound 12 as a white solid (0.68 g, 91 % yield). HPLC- MS tR = 2.13 min (UV254 nm); mass calculated for formula C13Hi8BrNO2299.1 , observed LCMS m/z 244.1 (M+H-'Bu). |
| 90% |
With sodium hydroxide; In 1,4-dioxane; water; at 0 - 20℃; for 5.5h; |
2-(4-Bromophenyl)-ethylamine (13, 10.0g, 50mmol) and NaOH (3.0g, 75mmol) were dissolved in the mixture of dioxane (150mL) and H2O (150mL). The reaction mixture was cooled to 0C. The solution of di-tert-butyl dicarbonate (16.36g, 75mmol) in dioxane (72mL) was added dropwise to the reaction mixture, which was stirred at room temperature for 5.5h. Dioxane was removed in vacuo and the residue was extracted with ethyl acetate 3 times. The combined organic layer was washed with water and brine, and then dried over anhydrous Na2SO4. Filtration and removal of solvent provided the crude product, which was purified by Combiflash chromatography (5-10% of ethyl acetate in hexane). The title compound 5 (13.5g, 90.0%) was obtained as a white solid; mp 59-60C. 1H NMR (300MHz, chloroform-d): delta=7.42 (d, J=8.0Hz, 2H), 7.07 (d, J=8.0Hz, 2H), 4.51 (br. s, 1H), 3.40-3.27 (m, 2H), 2.75 (t, J=6.9Hz, 2H), 1.43 (s, 9H). MS (EI): m/z=300.1 (M++H). |
| 90% |
With triethylamine; In dichloromethane; at 20℃; for 6h;Cooling with ice; |
Dissolve p-bromophenethylamine (2.0 g, 10 mmol) in a 100 mL round bottom flask under ice bathAfter dissolving 20 mL of dichloromethane, triethylamine (3.0 g, 30 mmol) was added, and then di-tert-butyl dicarbonate (3.3 g) was gradually added dropwise.20 mL of a solution of 15 mmol) of dichloromethane (DCM) was allowed to react at room temperature for 6 hours after the addition. After the reaction is completed, the solvent is distilled off under reduced pressure.40 mL of water was added, and the mixture was extracted with ethyl acetate. Distill the solvent and then silica gel column chromatographyFrom purification, eluent: petroleum ether (PE) / ethyl acetate (EA) = (20:1). A white solid (2.7 g, 90%) was obtained. |
| 89% |
With triethylamine; In dichloromethane; at 0 - 20℃; for 2h; |
To a stirred solution of 2-(4-bromophenyl)ethan-l -amine (3.0 g, 14.99 mmol) in dry DCM (30 ml) was added TEA (4.55 g, 44.98 mmol) at 0 C. Boc anhydride (3.92 g, (0366) 17.98 mmol) was added dropwise at 0 C and the resulting reaction mixture was allowed to come to room temperature over a period of 2 hours. The reaction mixture was poured into water (50 ml) and extracted with ethyl acetate (2 x 30 ml). The combined organic layers were washed with brine (50 ml), dried over anhydrous Na2S04 and concentrated under reduced pressure. The resulting residue was purified by silica gel chromatography to afford the title compound (4.0 g, 89 %) as off-white solid. 1H NMR (400 MHz, DMSO-d6): 1.36 (s, 9H), 2.66 (t, J = 7.2 Hz, 2H), 3.10-3.15 (m, 2H), 6.89 (S, 1H, -NH), 7.16 (d, J = 8.4Hz, 2H), 7.47 (d, J = 8.4Hz, 2H). LCMS: m/z = 244.1 [M+l -56] and 246.1 [M+2 -56]. |
| 88% |
|
To a solution of 4-bromophenethylamine (5.00 g, 25.0 mmol), potassium carbonate (5.18 g, 37.5 mmol) in CH2Cl2 (125 ml) was stirred for 15 min at room temperature, and boc anhydride was added (6.04 mL, 26.3 mmol). After 16 h, the reaction mixture was washed with H2O (125 mL 3) and brine (75 mL). The organic layer was dried with anhydrous Na2SO4 and concentrated in vacuo. The residue was washed with n-hexane and gave 13 as a white solid (6.63 g, 88%); Rf = 0.50 (EtOAc 1: n-Hexane 3); 1H NMR (300 MHz, DMSO-d6) delta 1.43 (s, C(CH3)3), 2.75 (t, J = 6.6 Hz, NHCH2CH2), 3.34-3.35(m, NHCH2CH2), 7.06 (d, J = 8.0 Hz, 2 ArH), 7.42 (d, J = 8.2 Hz, 2 ArH); 13C NMR (75 MHz, DMSO-d6) delta 28.4 (C(CH3)3), 35.7 (NHCH2CH2), 41.6 (NHCH2CH2), 79.3 (C(CH3)3), 120.2, 130.6, 131.6, 138.0 (6 ArC), 155.8 (C(O)). |
| 88% |
In tetrahydrofuran; at 0 - 20℃; for 18h; |
Di-te/f-butyl dicarbonate (3.93 g, 18.0 mmol) was added to a cooled (0C) stirred solution of 2-(4-bromophenyl)ethanamine (3.00 g, 15.0 mmol) in THF (20 ml). The resulting solution was allowed to warm to RT then stirred at RT for 18 h. The reaction mixture was partitioned between EtOAc (30 ml) and saturated aqueous NaHC03 solution (50 ml). The phases were separated then the aqueous phase was extracted with EtOAc (15 ml). The combined organic phases were washed with brine (50 ml), dried over MgS04, then concentrated in vacuo. The crude material was dissolved in the minimum volume of CH2CI2, pre-adsorbed onto silica, then purified by flash column chromatography on a silica column (25 g). The column was eluted with EtOAc: heptane, using the following gradient (%EtOAc, column volumes): 0%, 1 CV; 0-8%, 2.5 CV; 8-12%, 1 CV; 12%, 3.5 CV; 12- 27%, 5.5 CV; 27-30%, 0.5 CV; 30%, 2 CV; 30-90%, 4 CV; 100% 1 CV. The desired fractions were combined and concentrated in vacuo to afford the product as a white solid (3.99 g, 88%). 1 H NMR (500 MHz, DMSO-cfe) delta 7.50 - 7.42 (m, 2H), 7.19 - 7.1 1 (m, 2H), 6.86 (t, J = 5.3 Hz, 1 H), 3.12 (q, J = 6.6 Hz, 2H), 2.70 - 2.63 (m, 2H), 1.41 - 1.26 (m, 9H). LC/MS (System A): Rt = 1.27 min, UV purity = 99%. |
| 88% |
In tetrahydrofuran; at 0 - 20℃; for 18h; |
Di-teit-butyl dicarbonate (3.93 g, 18.0 mmol) was added to a cooled (0C) stirred solution of 2-(4-bromophenyl)ethanamine (3.00 g, 15.0 mmol) in THF (20 ml). The resultingsolution was allowed to warm to RT then stirred at RT for 18 h. The reaction mixture waspartitioned between EtOAc (30 ml) and saturated aqueous NaHCO3 solution (50 ml). The phases were separated then the aqueous phase was extracted with EtOAc (15 ml). The combined organic phases were washed with brine (50 ml), dried over Mg504, then concentrated in vacuo. The crude material was dissolved in the minimum volume of CH2CI2, pre-adsorbed onto silica, then purified by flash column chromatography on a silica column (25 g). The column was eluted with EtOAc:heptane, using the following gradient (%EtOAc, column volumes): 0%, 1 CV; 0-8%, 2.5 CV; 8-12%, 1 CV; 12%, 3.5CV; 12-27%, 5.5 CV; 27-30%, 0.5 CV; 30%, 2 CV; 30-90%, 4 CV; 100% 1 CV. Thedesired fractions were combined and concentrated in vacuo to afford the product as a white solid (3.99 g, 88%).1H NMR (500 MHz, DMSO-d6) O 7.50- 7.42 (m, 2H), 7.19-7.11 (m, 2H), 6.86 (t, J = 5.3Hz, 1H), 3.12 (q, J= 6.6 Hz, 2H), 2.70-2.63(m, 2H), 1.41- 1.26(m, 9H). LC/MS (System A): R = 1.27 mi UV purity = 99%. |
| 85% |
With N-ethyl-N,N-diisopropylamine; In dichloromethane; at 20℃; for 2h;Inert atmosphere; |
Intermediate 991 ,1 -Dimethylethyl [2-(4-bromophenyl)ethyl]carbamate Bis(1 ,1 -dimethylethyl) dicarbonate (240 mg, 1.100 mmol) was added to a mixture of [2-(4- bromophenyl)ethyl]amine 2-(4-bromophenyl)ethanamine (200 mg, 1 .000 mmol) and DIPEA (0.524 mL, 3.00 mmol) in dichloromethane (DCM) (5 mL). The resulting mixture was stirred under nitrogen at room temperature for 2 h then concentrated in vacuo. The residue was partitioned between AcOEt (20 mL) and water (15 mL) and the layers were separated. The aqueous phase was extracted with AcOEt (15 mL). The combined organic phases were washed with brine (15 mL), dried over MgS04 and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel (gradient: 5 to 25% AcOEt in hexanes) gave 1 ,1 -dimethylethyl [2-(4-bromophenyl)ethyl]carbamate (256mg, 0.853mmol, 85%) as a white solid.LCMS (method G): Retention time 1.22 min, [M+H-t-Bu]+ = 244.0 (1 Br) |
| 85% |
With N-ethyl-N,N-diisopropylamine; In dichloromethane; at 20℃; for 2h;Inert atmosphere; |
Bis(1,1-dimethylethyl) dicarbonate (240 mg, 1.100 mmol) was added to a mixture of [2-(4-bromophenyl)ethyl]amine 2-(4-bromophenyl)ethanamine (200 mg, 1.000 mmol) and DIPEA (0.524 mL, 3.00 mmol) in dichloromethane (DCM) (5 mL). The resulting mixture was stirred under nitrogen at room temperature for 2 h then concentrated in vacuo. The residue was partitioned between AcOEt (20 mL) and water (15 mL) and the layers were separated. The aqueous phase was extracted with AcOEt (15 mL). The combined organic phases were washed with brine (15 mL), dried over MgSO4 and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel (gradient: 5 to 25% AcOEt in hexanes) gave 1,1-dimethylethyl [2-(4-bromophenyl)ethyl]carbamate (256 mg, 0.853 mmol, 85%) as a white solid.LCMS (method G): Retention time 1.22 min, [M+H-t-Bu]+=244.0 (1 Br) |
| 84% |
In dichloromethane; at 25℃; for 4h; |
To a suspension of 2-(4-bromophenyl)ethan-1-amine (1) (3.1?mL, 20?mmol) in CH2Cl2 (30?mL) was added dropwise (Boc)2O (5.97?mL, 26?mmol) at 25?C and the solution was stirred for 4?h. After the reaction completed, the solvent was removed under reduced pressure. The residue was dissolved in water, then the aqueous solution was extracted with ethyl acetate (50?mL?*?3). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, and concentrated to give the crude product, which was purified by column chromatography to give tert-butyl(4-bromophenethyl)-carbamate (2) (5.0?g, 84% yield) as a white solid. 1H NMR (400?MHz, CDCl3) delta: 7.42 (d, J?=?8.2?Hz, 2H), 7.07 (d, J?=?8.1?Hz, 2H), 4.52 (s, 1H), 3.35 (s, 2H), 2.75 (t, J?=?6.9?Hz, 2H), 1.43 (s, 9H). |
| 65% |
With N-ethyl-N,N-diisopropylamine; In dichloromethane; at 20℃; for 4h;Inert atmosphere; |
[000449] Synthesis of tert-butyl (4-bromophenethyl) carbamate (547): To a stirred solution of 2-(4-bromophenyl) ethan-1-amine 546 (500 mg, 2.50 mmol) in CH2C12 (5 mL) under argon atmosphere were added Boc-anhydride (594 mg, 2.75 mmol), diisopropyl ethyl amine (1 mL, 7.50 mmol) at RT and stirred for 4 h. The reaction was monitored by TLC; after completion of the reaction, the reaction mixture was diluted with water (25 mL) and extracted with CH2C12 (235 mL). The combined organic extracts were washed with water (25 mL), dried over sodium sulfate, filtered and concentrated in vacuo to obtain the crude. The crude was purified through silica gel column chromatography using 5-8% EtOAc/ hexanes to afford compound 547 (500 mg, 65%) as white solid. TLC: 10% EtOAc/ hexanes (R 0.5); 1H-NMR (DMSO-d6, 400 MHz): oe 7.46 (d, J= 8.0 Hz, 2H), 7.12 (d, J= 8.0 Hz, 2H), 6.86-6.82 (m, 1H), 3.12-3.08 (m, 2H), 2.68-2.64 (m, 2H), 1.32 (s, 9H). |
| 53% |
|
Example 1 Synthesis of tert-butyl 4-bromophenethylcarbamate Bromophenethylamine (50 g, 250 mmol) and triethylamine (105 mL, 750 mmol) were stirred in dichloromethane (DCM; 1.5 L), and cooled to 0 C. Boc anhydride (82 g, 375 mmol) was added and the reaction mixture stirred at room temperature overnight. The reaction mixture was washed with water (1 L), brine (500 mL), dried (MgSO4) and concentrated to give an orange oil. The crude residue was crystallized from hexane (250 mL) to give a white solid, tert-butyl 4-bromophenethylcarbamate (39.85 g, 133 mmol, 53%). |
| 50% |
In tetrahydrofuran; at 20℃; for 1.5h; |
A solution of di-tert-butyl dicarbonate (0.36 g, 1.6 mmol) in tetrahydrofuran (5 mL) was added to a solution of 4-bromophenethylamine (0.30 g, 1.5 mmol) in tetrahydrofuran (10 mL), and the mixture was stirred at room temperature for 1.5 hours. After the reaction mixture was concentrated, the resultant solid was washed with hexane (3 mL), and dried under reduced pressure to give the title Reference compound (0.23 g) as a colorless solid (yield 50 %). tert-Butyl N-[2-(4-bromophenyl)ethyl]carbamate (Reference compound 30-1) [Show Image] 1H-NMR (500 MHz, CDCl3) delta 1.43 (s, 9H), 2.75 (t, J = 6.9 Hz, 2H), 3.3 5(q, J = 6.9 Hz, 2H), 4.52 (s, 1H), 7.07 (dd, J = 6.6, 1.7 Hz, 2H), 7.42 (dd, J = 6.6, 1.7 Hz, 2H) |
| 22% |
With dmap; In dichloromethane; at 20 - 24℃; for 18h; |
To a stirring solution of 4-dimethylaminopyridine (0.19 g, 1.5 mmol) and di-tert-butyl dicarbonate (2.6 g, 12 mmol) in dichloromethane (40 mL) was added 4-bromophenethylamine (1.6 mL, 10 mmol) and stirred at room temperature for 48 hours. The reaction mixture directly adsorbed onto silica gel and purified by flash column chromatography using 0-40% ethyl acetate/hexanes as eluent to afford the title compound as a white solid (0.68 g, 22%): mp 58-59 C.; 1H NMR (400 MHz, CDCl3) delta 7.42 (d, J=8.3 Hz, 2H), 7.07 (d, J=8.3 Hz, 2H), 4.51 (s, 1H), 3.43-3.27 (m, 2H), 2.75 (t, J=7.0 Hz, 2H), 1.43 (s, 9H); 13C NMR (101 MHz, CDCl3) delta 155.79, 137.97, 131.62, 130.55, 120.25, 77.21, 41.58, 35.65, 28.39; EIMS m/z 301 ([M]+). |
|
With triethylamine; In methanol; water; |
Step 1 A solution of 4-bromophenethylamine (10 g, 50 mmol), di-tert-butyldicarbonate (21.82 g, 100 mmol), triethylamine (13.9 ml, 100 mmol), and anhydrous methanol (350 ml) was refluxed for 2 h. The reaction mixture was concentrated in vacuo and the residue was stirred with water, filtered, washed with water and air dried to give N-(tert-butoxycarbonyl)-4-bromophenethylamine (15 g). |
|
|
EXAMPLE 63C 4-Bromo-(N-tert-butoxycarbonyl)phenethylamine 4-Bromophenethylamine and di-t-butyldicarbonate were subjected to the conditions described in Synthesis, 48, 1986 to provide the title compound. MS (DCI/NH3) m/e 319 (M+NH4)+. |
|
|
EXAMPLE 63C 4-Bromo-(N-tert-butoxycarbonyl)phenethylamine 4-Bromophenethylamine and di-t-butyldicarbonate were subjected to the conditions described in Synthesis, 48, 1986 to provide the title compound. MS (DCI/NH3) m/e 319 (M+NH4)+. |
|
In tetrahydrofuran; |
Preparation 74 Di-tert-butyl dicarbonate (2.18 g) was added to a solution of 2-(4-bromophenyl)ethanamine (2.0 g) in tetrahydrofuran (5 ml) under ice water cooling over 10 minutes and the mixture was stirred at room temperature for further 1 hour. The reaction mixture was evaporated in vacuo to give tert-butyl 2-(4-bromophenyl)ethylcarbamate (2.98 g) as a colorless foam. NMR (CDCl3, delta): 1.43 (9H, s), 2.75 (2H, t, J=8 Hz), 3.36 (2H, t, J=8 Hz), 7.00-7.10 (2H, m), 7.30-7.50 (2H, m) |
|
In tetrahydrofuran; for 0.166667h; |
To 7.03 g (35.1 mmol) of 4-bromophenethylamine (Sigma-Aldrich) in 60 mL of THF was added 8.6 g (39.4 mmol) of di-tert-butyldicarbonate. After 10 minutes, the solution was concentrated under reduced pressure, and the residue was partitioned between saturated aqueous sodium bicarbonate and ethyl acetate. The ethyl acetate phase was washed with brine, dried over MgSO4, filtered, and concentrated to give compound X as a white solid. |
|
With triethylamine; In methanol; for 2h;Heating / reflux; |
A solution of 4-bromophenethylamine (10 g, 50 mmol), di-tert-butyldicarbonate (21.82 g, 100 mmol), triethylamine (13.9 ml, 100 mmol), and anhydrous methanol (350 ml) was refluxed for 2 h.. The reaction mixture was concentrated in vacuo and the residue was stirred with water, filtered, washed with water and air dried to give N-(tert-butoxycarbonyl)-4-bromophenethylamine (15 g). |
|
With triethylamine; In DMF (N,N-dimethyl-formamide); at 50℃; for 0.25h; |
To a solution of 4-BROMOPHENETHYLAMINE (10G, 48. 98mmol) in anhydrous DMF (150mL), containing anhydrous triethylamine (35mL, 244.9 mol), was added BOC2O. The reaction mixture was heated for 15 minutes at 50C. After cooling to room temperature, brine (100ML) and HCI (1N, 100ML) were added subsequently, and the mixture was extracted several times with ether. The recombined organic layer was washed again with brine, dried over sodium sulfate, filtered and evaporated. The crude was flashed with 10% ethyl acetate in hexanes to give the [2- (4-BROMO-PHENYL)-ETHYL]-CARBAMIC acid tert-butyl ester.'H NMR (CDCI3, 300MHZ) : 1.41 (s, 9H); 2.22 (t, J=7. 1HZ, 2H); 3.31 (m, 2H); 4.67 (s, broad, 1 H); 7.03 (d, J=8.2Hz, 2H); 7.38 (d, J=8.2Hz, 2H). A mixture of the above mentioned compound (1. 00g, 3. 33MMOL), 3, 4-DIMETHOXYPHENYLBORONIC acid (1. 21 G, 6. 66MMOL), and potassium hydroxide (2N, 5mL, 10MMOL) in THF (15mL) was degassed using argon for 5 minutes. Palladium tetrakis-triphenylphosphine (200mg, 0. 167MMOL) was added and the mixture was heated at 85C. After 24 hours, the reaction mixture was allowed to cool to room temperature. Brine (20mL) was added and the reaction mixture was extracted several times with ether. The recombined organic phase was extracted with brine, dried over sodium sulfate, filtered, and evaporated. Silica gel chromatography of the crude using 20% ethyl acetate/n-hexane afforded [2- (3', 4'-DIMETHOXY-BIPHENYL-4YL)-ETHYLAMINE]-CARBAMIC acid tert-butyl ester as a white SOLID. 1H-NMR (CDCI3, 300MHZ) : 1.44 (s, 9H); 2.82 (t, J=6.9Hz, 2H); 3.39 (m, 2H); 3.91 (s, 3H); 3.93 (s, 3H); 4.67 (s, broad, 1H) ; 6.92 (d, J=8.2Hz, 1 H) ; 7.11 (m, 2H); 7.25 (m, 2H); 7.48 (d, J=8. 1 Hz, 2H). A solution of the above mentioned compound (1.14g, 3. 19mmol) in anhydrous methanol (50mL) was cooled in ice bath and then treated drop wise with acetyl chloride. Stirring was continued for 30 minutes at the same temperature followed by overnight stirring at room temperature. About 30mL of the solvent was removed by evaporation and the mixture was diluted with 200mL of ether. The entitled product was collected as a white solid by filtration, followed by washing with anhydrous ether and drying under high VACUUM.'H-NMR (D20, 300MHZ) : 2.80 (t, J=6.9Hz, 2H); 3.40 (m, 2H); 3.91 (s, 3H); 3.93 (s, 3H); 4.77 (s, broad, 1H) ; 7.00 (d, J=8.2Hz, 1H) ; 7.15 (m, 2H); 7.26 (m, 2H); 7.50 (d, J=8. 1HZ, 2H) |
|
With triethylamine; In dichloromethane; at 20℃; for 16h; |
Dissolved 2-(4-bromophenyl)ethanamine (25.0 g, 124.95 mmol) in DCM (500 mL). Charged with di-tert-butyl dicarbonate (32.7 g, 149.94 mmol) and triethylamine (52 mL, 373.08 mmol). Stirred at room temperature for 16 hours. Removed solvent and used crude without further purification. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping