| 79% |
|
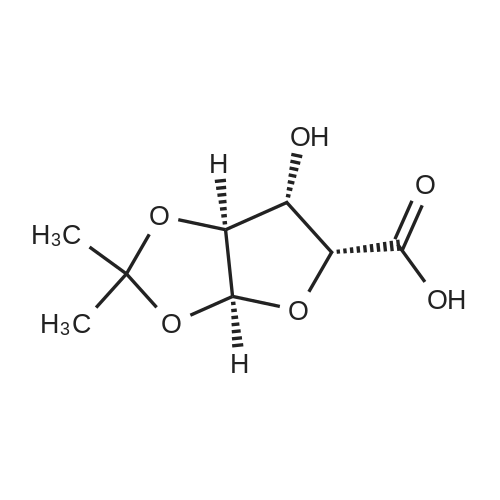
To a solution of (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol (25.0g, 131 mmol) in acetone (375 mL, 15X) and H2O (125 mL, 5X) was added NaHCO3 (33.0g, 3.0 equiv), NaBr (2.8g, 20 mol%) and TEMPO (0.40g, 2 mol%) at 20C. The mixture was cooled to 0-5C and solid trichloroisocyanuric acid (TCCA, 30.5 g, 1.0 equiv) was then added in portions. The suspension was stirred at 20C for 24h. Methanol (20 mL) was added and the mixture was stirred at 20C for 1h. A white suspension was formed at this point. The mixture was filtered, washed with acetone (50 mL, 2X). The organic solvent was removed under vacuum and the aqueous layer was extracted with EtOAc (300 mL, 12X x3) and the combined organic layers were concentrated to afford an oily mixture with some solid residue. Acetone (125 mL, 5X) was added and the mixture was filtered. The acetone solution was then concentrated to afford the desired acid ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid) as a yellow solid (21.0g, 79%). 1H NMR (methanol-d4), delta 6.00 (d, J = 3.2 Hz, 1H), 4.72 d, J = 3.2 Hz, 1H), 4.53 (d, J = 3.2 Hz, 1H), 4.38 (d, J = 3.2 Hz, 1H), 1.44 (s, 3H), 1.32 (s, 3H). |
| 77.2% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; trichloroisocyanuric acid; sodium hydrogencarbonate; sodium bromide; In methanol; water; acetone; at 0 - 30℃; for 25h; |
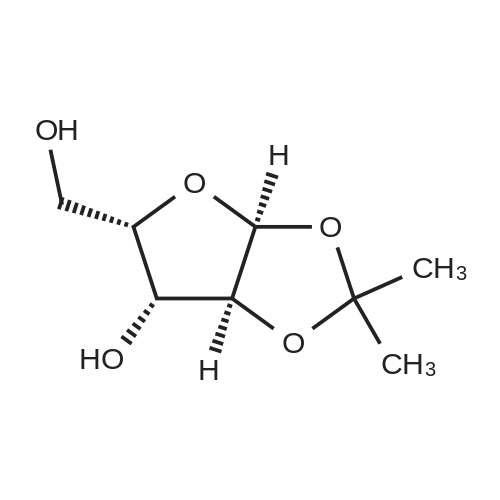
in room temperature,(3aS, 5S, 6R, 6aS) -5- (hydroxymethyl) -2,2-dimethyl-3a, 5,6,6a-tetrahydrofuro [2,3-d] [1,3] Oxepin-6-ol 1c (70.0 g, 368 mmol)Was dissolved in a mixed solvent of acetone and water (v / v = 5 / 2,1400 mL)Sodium bicarbonate (93 g, 1.11 mol),Sodium bromide (7.6 g, 74 mmol)And tetramethylpiperidine nitrogen oxide (1.2 g, 7.7 mmol) were successively added to the above solution.Cooling to 0 ,Sucralyl isocyanuric acid (86.0 g, 370 mmol) was added in portions,Stirred at room temperature for 24 hours,Then methanol (60 mL) was added,Stirring was continued for 1 hour.The solid was removed by filtration and washed with acetone (200 mL x 3). The filtrates were combined and the organic solvent was concentrated under reduced pressure. The residue was extracted with ethyl acetate (800 mL x 4). The solvent was concentrated under reduced pressure and acetone (400 mL) ,Filtration, vacuum concentration,The title compound was obtained as a reddish brown oil (58.0 g, 77.2%). |
| 58% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; trichloroisocyanuric acid; sodium hydrogencarbonate; sodium bromide; In N,N-dimethyl-formamide; at 20℃; for 12h; |
To a solution of ((3aS, 5S, 6R, 6aS) -5- (hydroxymethyl) -2,2-dimethyltetrahydrofuro [3,2- d] [1 Was added TEMPO (0.24 g, 1.5 mmol) to a solutionof 3-bromopyridin-3-yl] The mixture was cooled to 0 [deg.] C and then trichloroisocyanuric acid (17.8 g, 76.7 mmol) was added in portions. The suspensionwas stirred at room temperature for 12 hours. Methanol (2.0 mL) was added and themixture was stirred at room temperature for 2 hours. The mixture was filtered andwashed with acetone (2 x 20 mL wash). The organic solvent was removed in vacuo, theaqueous layer was extracted with EtOAc and the organic layer was concentrated invacuo. Acetone was added and the mixture was filtered. The filtrate was concentrated togive the title compound (9.0 g, 58%) as a light yellow solid. |
| 58% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; trichloroisocyanuric acid; sodium hydrogencarbonate; sodium bromide; In water; acetone; at 0 - 20℃; for 12h; |
To a solution of (178 (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,2-d][1,3]dioxol-6-ol (14.6 g, 76.7 mmol), 39 NaHCO3 (19.3 g, 230.3 mmol), and 180 NaBr (1.6 g, 15.4 mmol) in 42 acetone/9 water (120 mL/40 mL) was added 181 TEMPO (0.24 g, 1.5 mmol) at room temperature. The mixture was cooled to 0 C., and then 182 trichloroisocyanuric acid (17.8 g, 76.7 mmol) was added in small portions. The suspension was stirred at room temperature for 12 hours. 170 Methanol (2.0 mL) was added and the mixture was stirred at room temperature for 2 hours. The mixture was filtered and washed with acetone (twice, 20 mL per wash). The organic solvent was removed in vacuo, the aqueous layer was extracted with EtOAc, and the organic layer was concentrated in vacuo. Acetone was added thereto and the mixture was filtered. The filtrate was concentrated to obtain the desired 183 acid (9.0 g, 58%) as a light yellow solid. 1H NMR (400 MHz, CD3OD) delta 5.98 (d, J=3.6 Hz, 1H), 4.71 (d, J=3.2 Hz, 1H), 4.51 (d, J=3.6 Hz, 1H), 4.36 (d, J=3.6 Hz, 1H), 1.45 (s, 3H), 1.31 (s, 3H). |
|
With sodium hypochlorite; sodium chlorite; dipotassium hydrogenphosphate; potassium dihydrogenphosphate;2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; In water; acetonitrile; at 15 - 25℃;Industry scale;Product distribution / selectivity; |
A solution of the diol (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol in acetonitrile (5.38 kg, 65% w/w, 3.50 kg active, 18.40 mol), acetonitrile (10.5 L) and TEMPO (28.4 g, 1 mol %) were added to a solution of K2HPO4 (0.32 kg, 1.84 mol) and KH2PO4 (1.25 kg, 9.20 mol) in water (10.5 L). A solution of NaClO2 (3.12 kg, 80% w/w, 27.6 mole, 1.50 eq) in water (7.0 L) and a solution of K2HPO4 (2.89 kg, 0.90 eq) in water (3.0 L) were prepared with cooling. Bleach (3.0 L, approximate 6% household grade) was mixed with the K2HPO4 solution. Approximately 20% of the NaClO2 solution (1.6 L) and bleach/K2HPO4 solution (400 mL, 1 mol %) were added. The remainders of the two solutions were added simultaneously. The reaction mixture turned dark red brown and slow exotherm was observed. The addition rate of the NaClO2 solution was about 40 mL/min (3-4 h addition) and the addition rate for the bleach/K2HPO4 solution was about 10-12 mL/min (10 hr addition) while maintaining the batch at 15-25 C. Additional charges of TEMPO (14.3 g, 0.5 mol %) were performed every 5-6 hr until the reaction went to completion (usually two charges are sufficient). Nitrogen sweep of the headspace to a scrubber with aqueous was performed to keep the green-yellowish gas from accumulating in the vessel. The reaction mixture was cooled to <10 C. and quenched with Na2SO3 (1.4 kg, 0.6 eq) in three portions over 1 hr. The reaction mixture was then acidified with H3PO4 until pH reached 2.0-2.1 (2.5-2.7 L) at 5-15 C. The layers were separated and the aqueous layer was extracted with acetonitrile (10.5 L ×3). The combined organic layer was concentrated under vacuo (100-120 torr) at <35 C. (28-32 C. vapor, 45-50 C. bath) to low volume (6-7 L) and then flushed with acetonitrile (40 L) until KF of the solution reached <1% when diluted to volume of about 12-15Lwith acetonitrile. Morpholine (1.61 L, 18.4 mol, 1.0 eq) was added over 4-6 h and the slurry was aged overnight under nitrogen. The mixture was cooled to 0-5 C. and aged for 3 hours then filtered. The filter cake was washed with acetonitrile (10 L). Drying under flowing nitrogen gave 4.13 kg of the morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid as a white solid (92-94% pure based on 1H NMR with 1,4-dimethoxybenzene as the internal standard), 72-75% yield corrected for purity. 1H NMR (D2O) delta 5.96 (d, J=3.6 Hz, 1H), 4.58 (d, J=3.6 Hz, 1H), 4.53 (d, J=3.2 Hz, 1H), 4.30 (d, J=3.2 Hz, 1H), 3.84 (m, 2H), 3.18 (m, 2H), 1.40 (s, 1H), 1.25 (s, 1H). 13H NMR (D2O) delta 174.5, 112.5, 104.6, 84.2, 81.7, 75.0, 63.6, 43.1, 25.6, 25.1. |
|
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; trichloroisocyanuric acid; sodium hydrogencarbonate; sodium bromide; In water; acetone; at 0 - 20℃; for 24h; |
Step-Ill: To a stirred solution of (3aS,5S,6R,6aS)-5-hydroxymethyl-2,2-dimethyl- tetrahydro-furo[2,3-d][1 ,3]dioxol-6-ol (30 g, 157 mmol) in acetone (450 ml) and water (150 ml) was added sodium bicarbonate (39.8 g, 473 mmol), sodium bromide (3.25 g, 31 mmol) and TEMPO (490 mg, 3.1 mmol) at 20 C. The mixture was cooled to 0-5 C and solid trichloroisocyanuric acid (TCCA, 36.69 g, 157 mmol) was then added in portions. After stirring for 24 h at room temperature, methanol (25 ml) was added stirred for additional 1 h. The white suspension was formed. This was filtered, washed with acetone (50 ml X 2). The volatiles were evaporated under reduced pressure. The aqueous layer was extracted with ethyl acetate (300 ml X 3) and combined organic layers were concentrated to thick oily mixture with some solid residue. This was taken in acetone and filtered. The filtrate was concentrated to give 25 g of (3aS,5R,6S,6aS)-6- hydroxy-2,2-dimethyl-tetrahydro-furo[2,3-d][1 ,3]dioxole-5-carboxylic acid as a yellow solid. 1H NMR (400 MHz, CD3OD): delta 1.30 (s, 3H), 1.44 (s, 3H), 4.35 (d, J = 2.8 Hz, 1 H), 4.50 (d, J = 2.0 Hz, 1 H), 4.69 (d, J = 3.6 Hz, 1 H), 5.97 (d, J = 3.2 Hz, 1 H). |
|
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; [bis(acetoxy)iodo]benzene; In water; acetonitrile; at 30 - 50℃; |
Compound 3 (100 g, 525.8 mmol) was dissolved in a mixed solution of acetonitrile (380 mL) and water (190 mL), and 2,2,6,6-tetramethylpiperidine oxide (0.2-0.7 eq) was added.And iodobenzene diacetate (1.0-5.0 eq),The reaction mixture was stirred at 30-50 120-180 minutes, and extracted three times with ethyl acetate (500mL / times) and concentrated under reduced pressure to give a brown oily crude product (93.6g, 87%) ethyl acetate solution was concentrated. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping