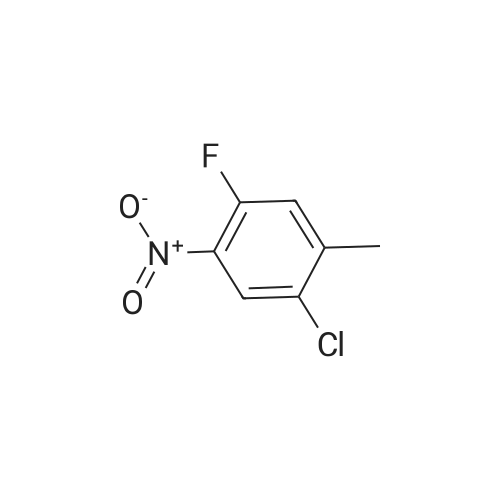
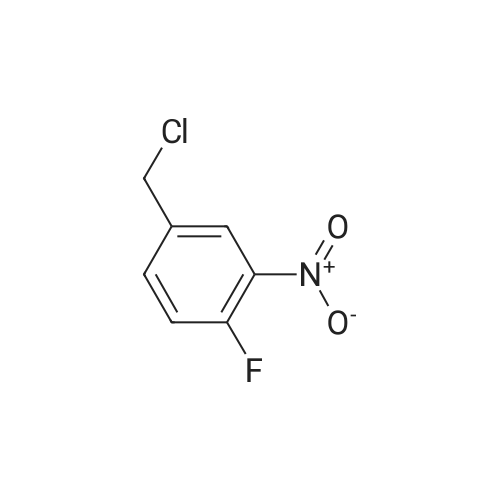
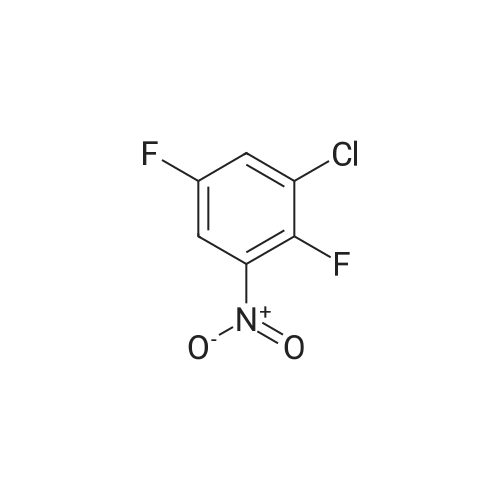
| 51.2% |
With sulfuric acid; potassium nitrate; at 0 - 20℃; for 22.0h; |
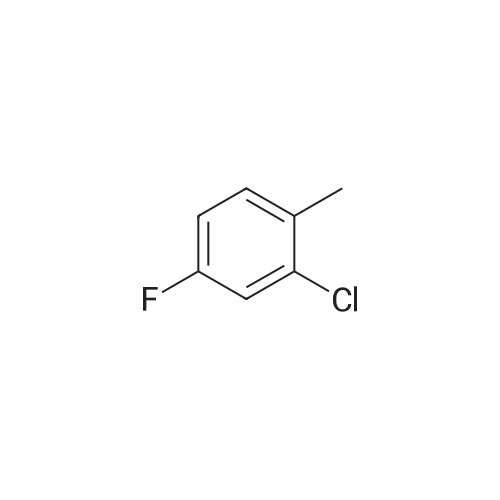
2-Chloro-4-fluoro-1-methylbenzene (5.000 g, 34.585 mmol) and Potassium nitrate (4.371 g, 43.232 mmol) was added to a mixture of sulfuric acid (12.536 mL, 235.180 mmol) at 0 & The mixture was slowly added so that the internal temperature did not exceed 10 C, slowly raised to room temperature, and stirred for 22 hours.The reaction mixture was cooled in an ice bath and poured into 50 ml of water. After completion of the reaction, the mixture was extracted three times with 50 ml of ethyl acetate.The combined organic layers were washed with a saturated aqueous sodium chloride solution, and water was removed with anhydrous magnesium sulfate, followed by filtration and concentration under reduced pressure.The concentrate was purified by column chromatography (SiO2, 80 g cartridge; ethyl acetate / hexane = 0% to 5%) and concentrated to give 1-chloro-5-fluoro-2-methyl- , 51.2%) as a pale yellow liquid. |
| 2.085 g (80%) |
With KNO3; In conc. H2 SO4; |
2-Chloro-4-fluoro-5-nitrotoluene. To a stirred solution of 2-chloro-4-fluorotoluene (2.000 g, 1.383 mmol, Aldrich, used as received) in conc. H2 SO4 (15.0 mL) at 0 C., KNO3 (1.400 g, 1.385 mmol) was added in one lot. The resulting pale yellow solution was allowed to warm to 28 C. and stirred overnight at 28 C. It was then poured into ice (100 g) and extracted with ethyl acetate (2*100 mL). Ethyl acetate was dried over anhydrous Na2 SO4, removed under vacuum, and the resulting oil was dried further under vacuum to afford 2.085 g (80%) of title compound as an oil, which was used as such for the next reaction; 1 H NMR (CDCl3): delta2.422 (s, 3H), 7.325 (d, 1H, J1 =10.2 Hz), 7.973 (d, 1H, J1 =5.2 Hz). |
|
With sulfuric acid; potassium nitrate; In 1,2-dichloro-ethane; |
Step A Synthesis of 2-chloro-4-fluoro-5-nitrotoluene Concentrated sulfuric acid (250 ml) was added to a stirred solution of 2-chloro-4-fluorotoluene (50.0 g, 0.346 mole) in ethylene dichloride (250 ml) at 0 C. Potassium nitrate (35.0 g, 0.346 mole) was then added slowly, maintaining the temperature below 10 C. The reaction was monitored by gas chromatography. Upon the disappearance of 2-chloro-4-fluorotoluene, the reaction mixture was poured into ice and extracted with methylene chloride. The dried organic layer was concentrated under reduced pressure to yield 60.0 g of 2-chloro-4-fluoro-5-nitrotoluene. This reaction was repeated several times. |
|
With sulfuric acid; potassium nitrate; In 1,2-dichloro-ethane; |
Step A Synthesis of 2-chloro-4-fluoro-5-nitrotoluene To a mixture of 20.0 g (0.138 mole) of 2-chloro-4fluorotoluene in 50 ml of 1,2-dichloroethane which had been cooled to 0 C. was added 50 ml of concentrated sulfuric acid. While maintaining the temperature below 10 C., 14.0 g (0.138 mole) of potassium nitrate was added slowly to the mixture. After three hours the reaction mixture was poured into ice, and the resulting mixture was extracted with methylene chloride. The extract was dried over anhydrous magnesium sulfate, filtered, and the solvent was evaporated under reduced pressure. The residue was passed through a silica gel column, eluding with heptane. Appropriate fractions were combined, and the solvent was evaporated under reduced pressure, yielding 13.0 g of 2-chloro-4-fluoro-5-nitrotoluene as a yellow solid. The nmr spectrum was consistent with the proposed structure. |
|
With sulfuric acid; potassium nitrate; at 0 - 20℃; for 1.0h; |
To a solution of 100 g (0.7 mol) of 2-chloro-4-fluorotoluene in 250 ml of concentrated H2SO4 is added portion-wise 85 g (0.875 mol) of KNO3 at 00C (addition of the whole amount of KNO3 is finished in about 1 hour). The reddish mixture is slowly warmed-up at room temperature overnight and quenched over crushed ice and extracted with EtOAc. The organic layers are combined, dried over MgSO4 and concentrated. The crude oil is then purified over a large silica plug (eluent: 97/3 hexanes/EtOAc) to afford 2-chloro-4-fluoro-5-nitrotoluene as a pale yellow oil that solidifies upon standing. 1H NMR (CDCl3, 400 Mz): 7.97 (d, J = 8.0 Hz, IH), 7.32 (d, J = 10.4 Hz, IH), 2.43 (s, 3H). |
| 1.1 g |
With sulfuric acid; potassium nitrate; at -5 - 20℃; for 16.0h; |
Step 1: 1-chloro-5-fluoro-2-methyl-4-nitrobenzene 2-chloro-4-fluoro-1-methyl benzene (1.5g, 10.4mmol) and concentrated sulfuric acid (15mL) were added into a 50 mL reaction flask. The reaction mixture was cooled down to -5C?0C and then potassium nitrate (1.4g, 13.8mmol) was added in batches at this temperature. The reaction mixture was slowly increased up to room temperature and stirred for 16 hours. After completion of the reaction, the reaction mixture was poured into ice water and extracted by using ethyl acetate, and washed with saturated aqueous sodium bicarbonate and saturated brine, dried and concentrated. The crude product thus obtained was separated and purified by column chromatography (silica gel column, eluent: ethyl acetate / petroleum ether = 1:50) to obtain the title compound (yellow solid, 1.1g, 56%). (MS: [M+1] none) |
|
With sulfuric acid; nitric acid;Cooling with ice; |
135ml concentrated sulfuric acid into the reaction bottle, Stirring under ice bath cooling, 43.4 g of fuming nitric acid was added dropwise. After dripping continue to stir 30min, Form mixed acid. And 315 ml of concentrated sulfuric acid (3.5 V) and 90.0 g of 2-chloro-4-fluoro-toluene were charged in another three-necked flask. Ice salt bath cooling, The mixed acid of nitric acid and sulfuric acid is added to the sulfuric acid solution of 2-chloro-4-fluoro-toluene, Continue to react 1 ~ 2 hours. The reaction solution was slowly added to the crushed ice to stir, Ethyl acetate was extracted twice. Combined with ethyl acetate, Washed twice with water, Saturated brine once. Organic phase concentrated to dry, To give 2-chloro-4-fluoro-5-nitro-toluene as an oil. |
|
With sulfuric acid; nitric acid;Cooling with ice; |
EXAMPLE 1 Preparation of 2-chloro-4-fluoro-5-nitrotoluene 135 ml of concentrated sulfuric acid was added into the reaction flask, and cooled in an ice bath under stirring, 43.4g fuming nitric acid was then added therein dropwise. After the completion of addition, the mixture was stirred continuously for 30 min to form a mixed acid. Meanwhile, 315 ml of concentrated sulfuric acid and 90.0 g of 2-chloro-4-fluorotoluene were added in another three-necked flask. The abovementioned mixed acid of nitric acid and sulfuric acid was added to the sulfuric acid solution of 2-chloro-4-fluorotoluene under cooling in an ice-salt bath, and the reaction was continued for 1 to 2 hours. The reaction solution was slowly added to the crushed ice under stirring to quench, and then extracted with ethyl acetate twice. The ethyl acetate layers was combined, and washed twice with water and once with saturated brine. The organic phase was concentrated to dryness to give oily 2-chloro-4-fluoro-5-nitrotoluene. H NMR Data 1H NMR(CDCl3, 400 MHz): delta 7.95(d, 3.8, 1H), 7.51(d, 4.2, 1H), 2.45(s, 3H). |
|
With sulfuric acid; potassium nitrate; at 20℃; for 1.0h; |
Description 84. 1-Chloro-5-fluoro-2-methyl-4-nitrobenzene (D84); A solution of 2-chloro-4-fluorotoluene (4.Og) in concentrated sulfuric acid (30ml) was treated portionwise with potassium nitrate (2.8g) at <20C (internal). After 1 h more stirring with ice cooling the mixture was poured onto ice and extracted with diethyl ether. Drying and evaporation gave the title compound, 4.5g. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping