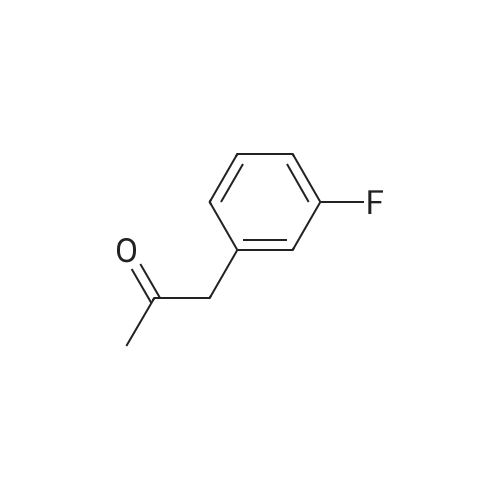
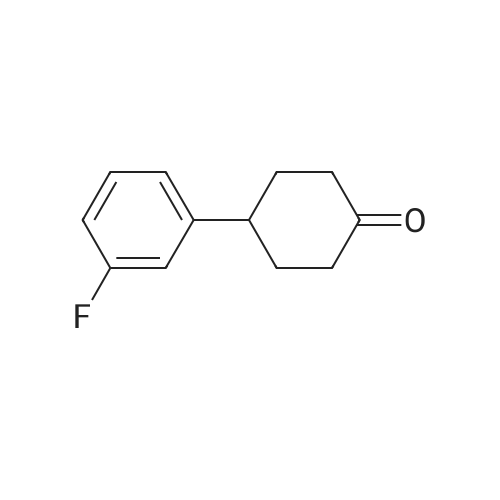
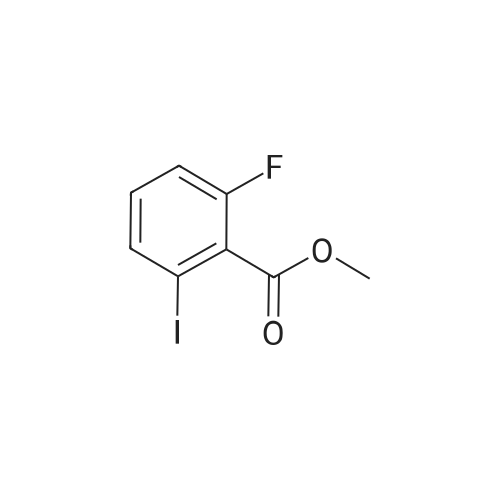
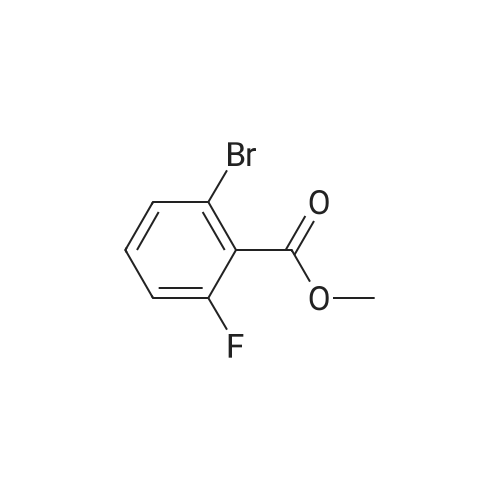
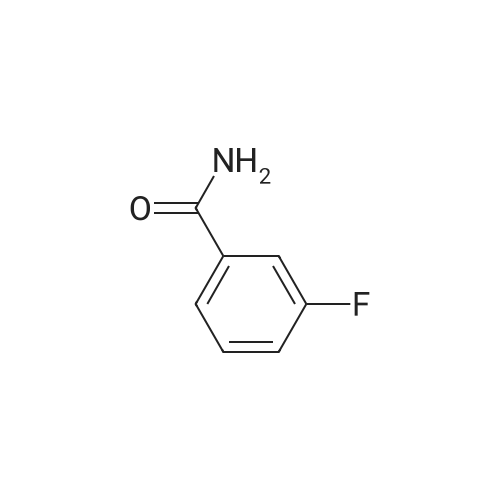
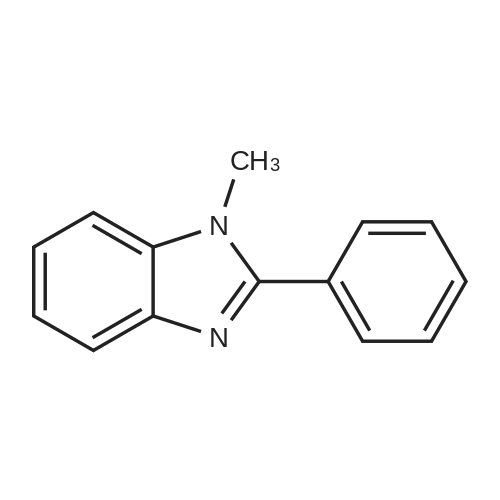
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
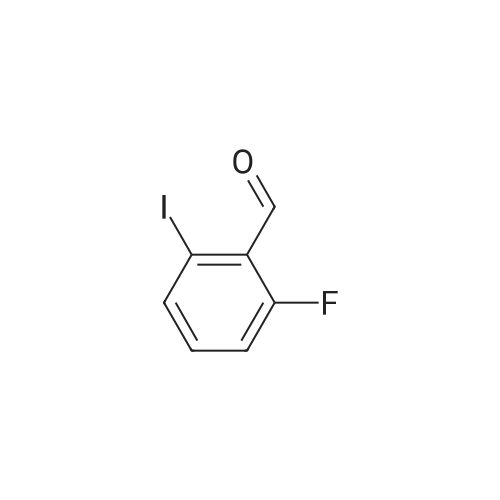
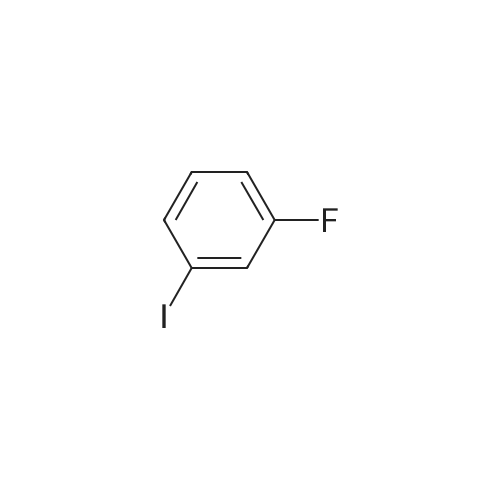
Step:1 To a solution of l -fluoro-3-iodobenzene (5.00 g, 22.5 mmol) in THF (50 mL), was added lithium diisopropylamide (17.0 mL, 33.7 mmol) dropwise at -78 C. After being stirred at -78 C for 2 hours, N N-dimethylformamide (4.90 g, 67.5 mmol) was added and the resulting mixture was stirred at -78 C for another 30 min. The reaction mixture was then treated with aq. solution of ammonium chloride (20 mL) and water (30 mL), extracted with diethyl ether (3 x 30 mL). The combined organic layers were washed with 2 Nu hydrochloric acid (20 mL) and brine (20 mL), dried over anhydrous sodium sulfate. The solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel eluting with petroleum/ethyl acetate (100:1 to 50:1) to give the desired product (3.7 g, 66% yield). lU NMR (DMSO-i? , 500 MHz): delta 10.01 (s, 1H), 7.89 - 7.79 (m, 1H), 7.44 - 7.40 (m, 2H).
66%
Step 1: To a solution of 1 -fluoro-3-iodobenzene (5.00 g, 22.5 mmol) in THF (50 mE), was added lithium diisopropylamide (17.0 mE, 33.7 mmol) dropwise at -78 C. After being stirred at -78 C. for 2 hours, N,N-dimethylformamide (4.90 g, 67.5 mmol) was added and the resulting mixture was stirred at -78 C. for another 30 mm. The reaction mixture was then treated with aq. solution of ammonium chloride (20 mE) and water (30 mE), extracted with diethyl ether (3x30 mE). The combined organic layers were washed with 2 N hydrochloric acid (20 mE) and brine (20 mE), dried over anhydrous sodium sulfate. The solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel eluting with petroleuml ethyl acetate (100:1 to 50:1) to give the desired product (3.7 g, 66%yield). 1HNMR(DMSO-d5, 500MHz): oe 10.01 (s, 1H),7.89-7.79 (m, 1H), 7.44-7.40 (m, 2H).
Buthyllithium (1.6 M, 60 ml, 0.113 mol) is added to an ice cooled solution of diisopropyl- amine (17.6 ml, 0.124 mol) in THF (200 ML). The solution is stirred at 0C for 30 minutes and is cooled TO-78 OC. I-FLUORO-3-IODO-BENZENE (25 g, 0.113 mol) in THF (20 ML) is slowly added to the preformed LDA mixture. The resulting solution is stirred for another hour after which DMF (9.55 ml, 0.124 mol) is added and stirring is continued for another 30 minutes at - 78 C. The reaction is quenched first with acetic acid (20 ml) followed by water (200 ML). The mixture is extracted with ether. The organic layer is washed with a 2N HCI solution, brine, is dried over MGS04 and evaporated to afford the desired compound (28.3 g).
General procedure: To a stirred solution of 1-fluoro-2-iodobenzene (4.6 mL, 39 mmol) in THF (39 mL)at -78C was added 1.8 M LDA solution (22.8 mL, 1.05 equiv.) dropwise over 10 minunder Ar. After 10 min, DMF (3.65 mL, 1.2 equiv.) was added dropwise and the reactionwas stirred for 10 min at -78C. The reaction was quenched at -78C by the rapid additionof 10 mL of acetic acid followed by 80 mL of H2O. The cooling bath was removed, andthe aqueous phase was extracted with diethyl ether (70 mL x 3). The organic layers werecombined and washed with 0.5 N HCl solution (150 mL x 2), H2O (150 mL x 2), and brine(150 mL). The organic solution was dried over MgSO4, filtered, and concentrated underreduced pressure. The resulting brown solid (6.25 g, 64% crude yield) was dried in vacuoand used for the next reaction without further purification.
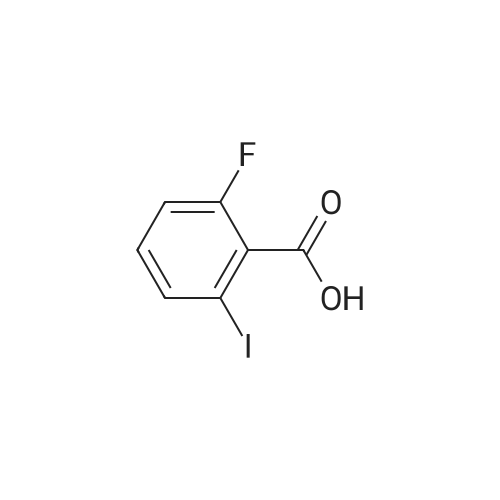
n-BuLi (120 rnL) was added dropwise to a solution of diisopropylamine (33.33 g, 330.00 mmol) in ether (300 rnL) at -700C. The resulting solution was stirred at -700C for 1 h followed by addition of a solution of l-fluoro-3-iodobenzene (22.2 g, 100.00 mmol) in ether (100 mL). After stirring for 1 h (at -700C), CO2 (gas) was bubbled in the reaction mixture. The resulting solution was stirred at -500C for 1 h. The resulting solution was extracted with water (1x300 mL). Adjustment of the pH to 1 was accomplished by the addition of HCl (4M) and the aqueous layer was extracted with EtOAc (3x300 mL). The organics were combined, washed with brine (2x100 mL), dried over Na2SO4, and evaporated to dryness to afford 8 g (33%) of 2-fluoro-6- iodobenzoic acid as a yellow solid.
To a solution of diisopropylamine (6.94 ml, 49.5 mmol, 1.1 eq.) in THF (90 ml) under N2, stirring at 0-5 C., was added dropwise over 10 minutes n-BuLi (28.12 ml, 45 mmol) and stirred at this temperature for 10 min. The reaction mixture was cooled to -78 C. in a dry ice/acetone bath and 1-iodo-5-fluorophenyl (5.29 ml, 45 mmol) was added dropwise over 5 min. The reaction was stirred at this temperature for 1 h, then dropwise DMF (4.15 ml, 49.5 ml, 1.1 eq.) was added over 5 minutes, and stirred for 10 minutes. Acetic acid (9 ml) was added, followed by H2O, and extracted with Et2O. The combined organic solution was washed with 0.1 N HCl, brine, dried (MgSO4), filtered and concentrated. The crude 2-iodo-6-fluorobenzaldehyde (9.65 g) was taken onto the next step without purification. 1H NMR (400 MHz, CDCl3) delta 10.13 (s, 1H), 7.80 (d, 1H), 7.31-7.21 (m, 2H). [00422] GC[100 C (5 min.)?180 C (5 min.) 20 C/min], Rt=8.067.
With 1,4-diaza-bicyclo[2.2.2]octane; N-methoxylamine hydrochloride; sodium iodide; palladium dichloride; In acetonitrile; at 90℃; under 3800.26 Torr; for 8h;Autoclave; Inert atmosphere;
General procedure: To an autoclave (100 mL capacity), were added an arylhalide (1 mmol), methoxylamine hydrochloride (1.2 equiv),DABCO (2 equiv), PdCl2 (10 mol%), NaI (0.2 mmol) and MeCN (15 mL), under an inert atm. The autoclave was flushed three times with CO and then pressurized to 5 atm of CO. The mixture was stirred with a mechanical stirrer (550rpm) at 90 C for 8 h. The reactor was cooled to r.t., degassed carefully, opened and the reaction mixture removed. The reactor vessel was washed with EtOAc (2 × 5 mL) to remove residual product. The mixture was filtered and the filtrate washed with brine (2 × 4 mL), dried over Na2SO4, filtered and the solvent evaporated under vacuum. Purification of the residue was carried out by column chromatography (silicagel, 100-200 mesh, PE-EtOAc) to afford the corresponding product in good to excellent yield.
2-(3'-fluorobiphenyl-2-yl)-1-methyl-1H-benzoimidazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
82%
With [ruthenium(II)(eta6-1-methyl-4-isopropyl-benzene)(chloride)(mu-chloride)]2; potassium carbonate; triphenylphosphine; In benzene; at 150℃; for 24h;Sealed tube;
General procedure: In a 30-mL sealed tube, <strong>[2622-63-1]1-methyl-2-phenylbenzimidazole</strong> (1, 0.25mmol), [RuCl2(p-cymene)]2 (0.0125 mmol), Ph3P (0.075 mmol),K2CO3 (0.50 mmol), and iodoarene (0.25 mmol) were combined inanhydrous benzene (2 mL) under air. The mixture was then stirredat 150 °C for 24 h. The mixture was cooled to r.t., diluted with EtOAc, and filtered through a small pad of Celite. The filtrate was concentrated in vacuo and purified by flash chromatography (silicagel, EtOAc?hexane) to give the analytically pure 2-(biphenyl-2-yl)benzimidazoles.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping