| 88% |
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 2 h; Inert atmosphere
Stage #2: With Trimethyl borate In tetrahydrofuran; hexane at -78 - 20℃; for 20 h;
Stage #3: With hydrogenchloride In tetrahydrofuran; hexane; water for 7 h; |
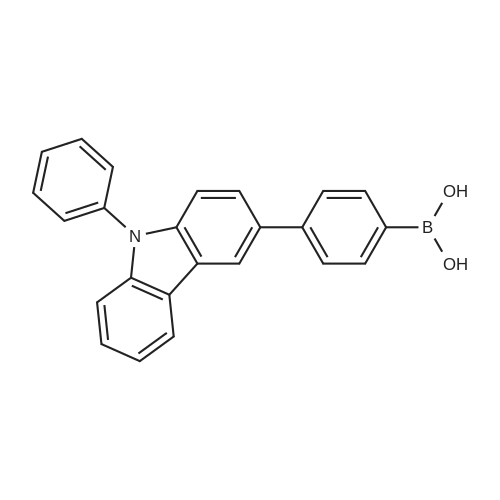
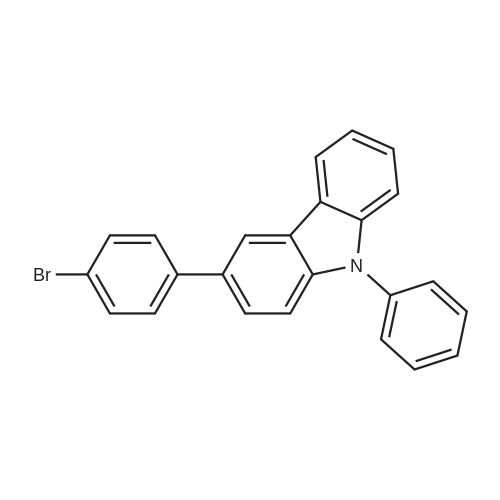
Step 2: Synthesis Method of 4-(9-phenyl-9-H-carbazol-3-yl)phenylboronic acidA synthetic scheme for 4-(9-phenyl-9-H-carbazol-3-yl)phenylboronic acid is illustrated in the following (F-2). Into a 300-mL three-neck flask, 8.0 g (20 mmol) of the 3-(4-bromophenyl)-9-phenyl-9H-carbazole obtained in Step 1 described above was put, the atmosphere in the flask was replaced with nitrogen, 100 mL of dehydrated tetrahydrofuran (abbreviation: THF) was then added to the flask, and the temperature was lowered to -78° C. Into this mixture solution, 15 mL (24 mmol) of a 1.65 mol/L n-butyllithium hexane solution was dropped, and the mixture solution with the n-butyllithium hexane solution added was stirred for 2 hours. To this mixture, 3.4 mL (30 mmol) of trimethyl borate was added, and the mixture with the trimethyl borate added was stirred at -78° C. for 2 hours and at room temperature for 18 hours. After the reaction, 1M diluted hydrochloric acid was added to this reaction solution until the solution became acid, and the solution with the diluted hydrochloric acid added was stirred for 7 hours. This solution was subjected to ethyl acetate extraction, and the obtained organic layer was washed with a saturated saline. After the washing, magnesium sulfate was added to the organic layer to adsorb moisture. This suspension was filtrated, and the obtained filtrate was concentrated, and hexane was added thereto. The mixture was exposed to supersonic waves and then recrystallized to obtain an intended white powder with a yield of 6.4 g at 88percent.The Rf value of the intended product obtained by silica gel thin layer chromatography (TLC) (developing solvent, ethyl acetate:hexane=1:10) was 0 (origin), and the Rf value of the 3-(4-bromophenyl)-9-phenyl-9H-carbazole was 0.53. In addition, the Rf value of the intended product obtained by silica gel thin layer chromatography (TLC) using ethyl acetate as the developing solvent was 0.72, and the Rf value of the 3-(4-bromophenyl)-9-phenyl-9H-carbazole was 0.93. In any case of the developing solvents, no spot derived from the 3-(4-bromophenyl)-9-phenyl-9H-carbazole was observed. Therefore, it is determined that the implementation of the reaction in Step 2 provided the intended product with a higher degree of purity in a simple manner with an extremely high yield.In Step 2, the 3-(4-bromophenyl)-9-phenyl-9H-carbazole as a raw material halide was reacted with n-butyllithium whose amount (1.2 equivalents) is larger than that of the 3-(4-bromophenyl)-9-phenyl-9H-carbazole as a lithiating agent so that the raw material halide was not left. In addition, the raw material halide is more likely to dissolve in a nonpolar solvent such as hexane, than the boron compound (4-(9-phenyl-9-H-carbazol-3-yl)phenylboronic acid) as the intended product, and can be thus easily separated by recrystallization. This allows for preventing an impurity from being produced by a coupling reaction between the boron compound (4-(9-phenyl-9-H-carbazol-3-yl)phenylboronic acid) synthesized in Step 2 and a halide (3-(4-bromophenyl)-9-phenyl-9H-carbazole), which was an impurity in the boron compound, in the next step of a reaction with a halogenated anthracene compound. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping