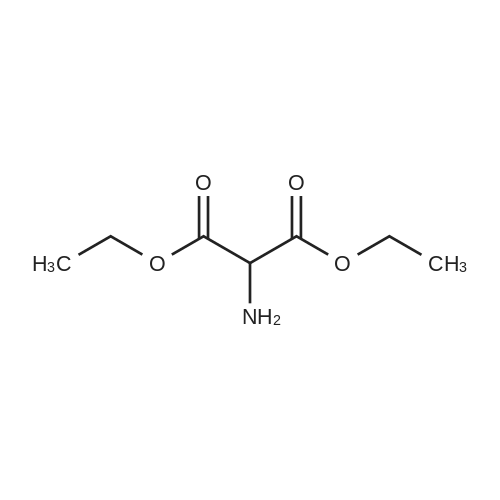
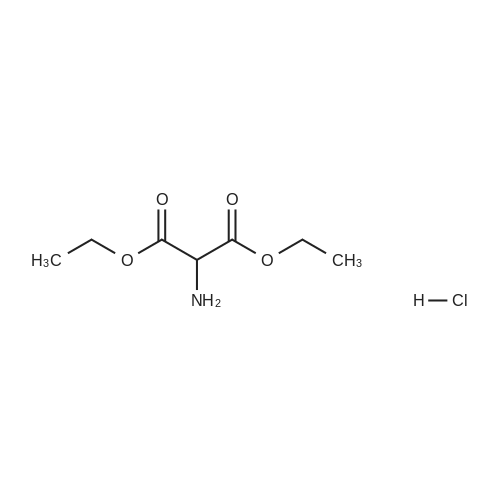
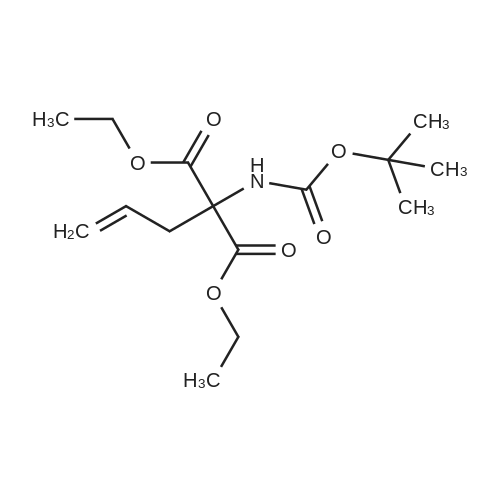
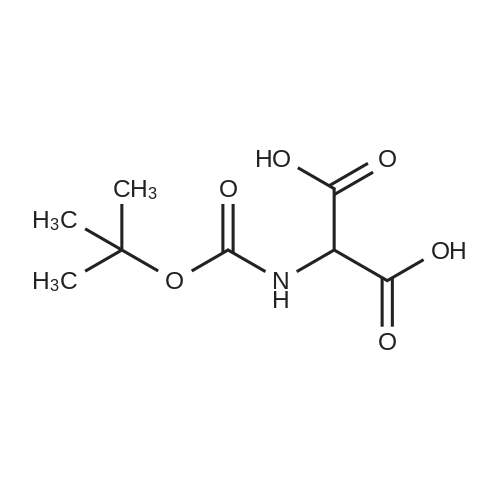
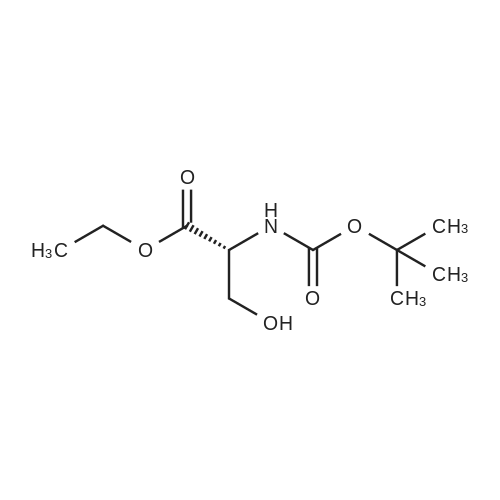
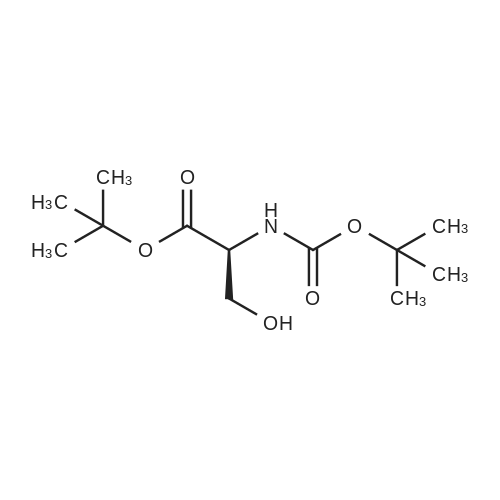
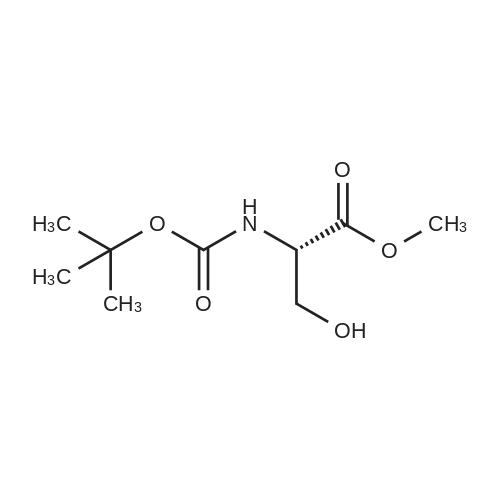
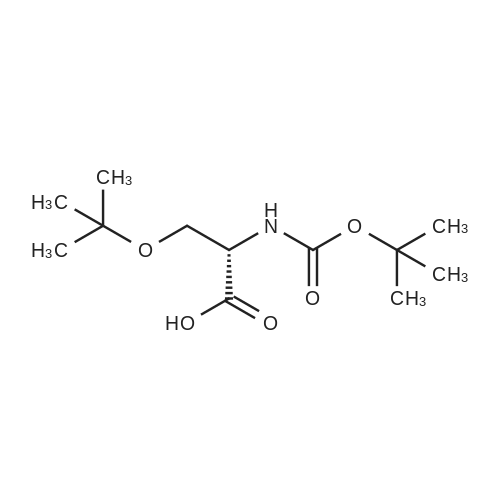
| 91% |
|
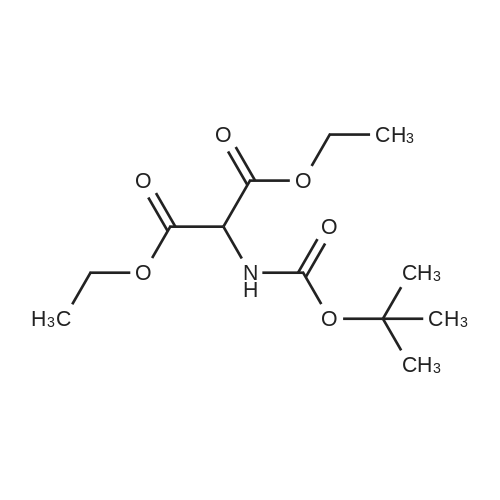
This compound was synthetized according to the procedure describedby Berner and co-workers.21 To a solution of 10.0 g of diethylaminomalonate hydrochloride (47.3 mmol, 1.008 equiv) in 80.0 mLof dioxane, 1.89 g of NaOH in water (1.01 equiv, 47.4 mmol, 1.0 M)were added. After complete dissolution of the salt, a solution of10.3 g of (Boc)2O (1.0 equiv, 46.8 mmol) in 20.0 mL of dioxane wasadded dropwise and reacted overnight. Once the reaction wasfinished the solvents were removed at reduced pressure, the crudesolid was redissolved with EtOAc, washed with solutions of 1 N HCland saturated NaCl, dried over MgSO4 anhyd and the solvent of thecombined organic phases was removed at reduced pressure. Finally,the crude product was purified by flash column chromatography(Hexane/EtOAc 3:1) to afford the desired product as a whitesolid in 91% yield. 1H NMR (400 MHz, CDCl3) d5.54 (br s, 1H), 4.93(d, J7.8 Hz, 1H), 4.35e4.15 (m, 4H), 1.44 (s, 9H), 1.29 (t, J7.1 Hz,6H). |
| 91% |
|
Diethyl 2-aminomalonate hydrochloride (2.54 g, 12.0 mmol) was dissolved in a mixture of 1M NaOH (12 mL) and 1,4-dioxane (10 mL) and a solution of Boc-anhydride (2.54 g, 12.0 mmol, 1.0e q.) in 1,4-dioxane (5 mL) was added dropwise at 5C. Subsequently, the mixture was stirred at r.t. for 24 h. Dioxane was removed in vacuo and the residue was dissolved in ethyl acetate. After phase separation, the organic layer was washed with 1M HCl (3×50mL) and dried over Na2SO4. The solvent was removed in vacuo and the crude product was purified by column chromatography (cyclohexane/ethyl acetate, 6:1). The product was isolated as colorless oil. Yield: 3.01g (91%). 1H NMR (300 MHz, CDCl3): δ 1.30 (t, 3JH,H=7.2Hz, 6H, 10-CH3, 12-CH3), 1.45 (s, 9H, 6-CH3, 7-CH3, 8-CH3), 4.27 (m, 4H, 9-CH2, 11-CH2), 4.94 (d, 3JH,H=7.7Hz, 1H, 2-CH), 5.63 (d, 3JH,H=7.8Hz, 1H, 2-NH).113C NMR (101 MHz, CDCl3): δ 14.0 (q, C-10, C-12), 28.2 (q, C-6, C-7, C-8), 57.5 (d, C-2), 62.4 (t, C-9, C-11), 80.5 (s, C-5), 154.8 (s, C-4), 166.6 (s, C-1, C-3).Exact mass (ESI+): C12H21NO6 + Na+: calcd. 298.1261, found 298.1244. |
| 89% |
|
Compound 1 (500g, 2.36mol) was dissolved in 1L of methylene chloride, added dropwise triethylamine (716g, 7.08mol) under ice-water bath, the addition was complete the reaction at room temperature 0.5h; cooled in an ice-water bath, slowly added Boc anhydride (567g, 2.60mol), completion, stirring overnight at room temperature.TLC showed disappearance of compound 1, the reaction was filtered, the filter cake was washed with dichloromethane, and the filtrate was concentrated to give an oil.Oil was dissolved in ethyl acetate was added, washed with 1N aqueous hydrochloric acid solution, then with saturated brine, the organic phase was dried over anhydrous sodium sulfate, filtered, the organic phase was concentrated to give 578g of Compound 2 as a yellow oil (yield: 89%), used without further purification in the next reaction. |
| 85% |
With sodium hydroxide; In water; acetone; for 48h;pH 8; |
Step 1 To a solution of diethyl aminomalonate hydrochloride XCV (2.0 g, 9.45 mmol) in water (45 mL) was added 1 M NaOH to pH~8. Boc2O (3.72 g, 17.0 mmol) in acetone (15 mL) was then added. The reaction mixture was stirred for 2 days before the acetone was evaporated under reduced pressure. The residue was washed by diethyl ether, and the organic layer was evaporated under vacuum to give the crude 1,3-diethyl 2-[(tert-butoxy)carbonyl]amino}propanedioate XCVI as a colorless oil (2.22 g, 8 mmol, 85% yield). The crude product was used directly in step 2. ESIMS found for C12H21NO6 m/z 276 (M+H). |
| 46% |
|
NaHCO3 (462 mg, 5.5 mmol)was slowly added to a suspension ofdiethyl aminomalonate hydrochloride(1.0582 g, 5 mmol, 13) in water (7 mL) and dioxane(10 mL). The resulting solution was stirred for a fewminutes at room temperature (rt) until a clear solutionappeared. Next, DMAP (6.11 mg, 0.01 mmol) was addedfollowed by a dropwise addition of a solution of Boc2O(1.2004 g, 5.5 mmol) in dioxane (4 mL). The mixture wasstirred at room temperature overnight. Then, the solutionwas concentrated under reduced pressure. The residue wassuspended in ethyl acetate (25 mL) and then extracted with5% aqueous KHSO4 solution (20 mL), saturated aqueousNaHCO3 solution (20 mL), water (15 mL) and brine (15 mL).The organic phase was dried with anhydrous Na2SO4 andconcentrated under reduced pressure. 1.2803 g, 4.65 mmol(46% yield) of a light oil was obtained and applied for thenext reaction without further purification. LRMS (ESI(electrospray ionization)) m/z 276.1 [M - CO2]+, 100). |
|
With triethylamine; In tetrahydrofuran-water; |
(a) A solution of 31.7 g (150 mmole) of diethyl aminomalonate hydrochloride in 400 mL 1/1 THF/H2O was cooled to 0C and 20 mL triethylamine was added followed by 35 g di-t-butyldicarbonate. The reaction mixture was stirred 1.25 hours at 0C and was then warmed briefly to 50C. The reaction mixture was stirred overnight at room temperature, concentrated to remove THF, diluted with ethyl acetate, washed with 200 mL 10% citric acid solution and brine, dried (MgSO4), filtered, and concentrated to a clear viscous liquid to give N-[bis(ethoxycarbonyl)methyl]carbamic acid, 1,1-dimethylethyl ester. 43.3 g, 100%. IR (LF) 2938, 1761, 1720, 1506, 1370, 1163, 780 cmmin1. |
|
With N-ethyl-N,N-diisopropylamine; In dichloromethane; at 20℃; for 16h;Cooling with ice; |
Step I. To a solution of diethyl aminomalonate hydrochloride (17 g, 80 mmol) and DIPEA (44 mL, 250 mmol) in DCM (250 mL) was added di-tert-butyl dicarbonate (20.8 g, 96 mmol) at ice-bath. The mixture was allowed to warm to room temperature while stirred for 16 h. Then the mixture was concentrated in vacuo. And the residue was extracted with DCM (150 mL) two times. The combined organic layers were washed with saturated NaHCO3 (100 mL) three times, dried over anhydrous sodium sulfate and concentrated in vacuo to afford 22 g of crude product diethyl 2-(tert-butoxycarbonylamino)propanedioate as a colorless oil (yield was 100%). MS: calc'd (MH+) 276, measured (MH+) 276 |
|
With N-ethyl-N,N-diisopropylamine; In dichloromethane; at 20℃; for 16h;Cooling with ice; |
To a solution of diethyl aminomalonate hydrochloride (17 g, 80 mmol) and DIPEA (44 mL, 250 mmol) in DCM (250 mL) was added di-tert-butyl dicarbonate (20.8 g, 96 mmol) at ice-bath. The mixture was allowed to warm to room temperature while stirred for 16 h. Then the mixture was concentrated in vacuo. And the residue was extracted with DCM (150 mL) two times. The combined organic layers were washed with saturated NaHC03 (lOOmL) three times, dried over anhydrous sodium sulfate and concentrated in vacuo to afford 22 g of crude product diethyl 2-(tert-butoxycarbonylamino)propanedioate as a colorless oil (yield was 100%). MS: calc'd (MH+) 276, measured (MH+) 276. |
|
With dmap; sodium hydrogencarbonate; In 1,4-dioxane; water; at 20℃; for 12h; |
To a solution of diethyl 2-aminomalonate hydrochloride (50 g, 236 mmol) in H2O (300 mL) and dioxane (440 mL) was slowly added NaHCO3 (21 g, 248 mmol) at 20 C. When the solution became clear, DMAP (289 mg, 2 mmol) was added followed by dropwise addition of a solution of Boc2O (54 g, 248 mmol) in dioxane (160 mL).The mixture was stirred at 20 C. for 12 hours. The mixture were concentrated. The residue was dissolved in ethyl acetate. The organic phase was washed with solution of 5% KHSO4 (aq.), sat. aq. NaHCO3, water, and brine, and dried over anhydrous Na2SO4, then filtered and concentrated to give diethyl 2-((tert-butoxycarbonyl)amino)malonate. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping