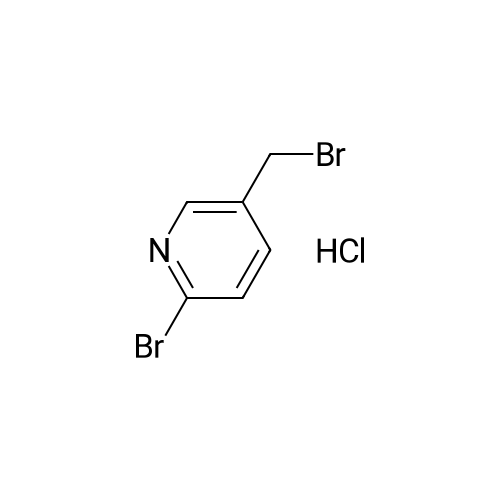
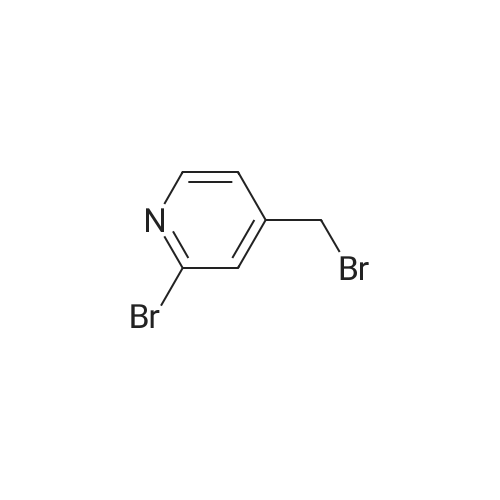
| 59% |
With N-Bromosuccinimide; dibenzoyl peroxide; In Carbon tetrachloride;Reflux; |
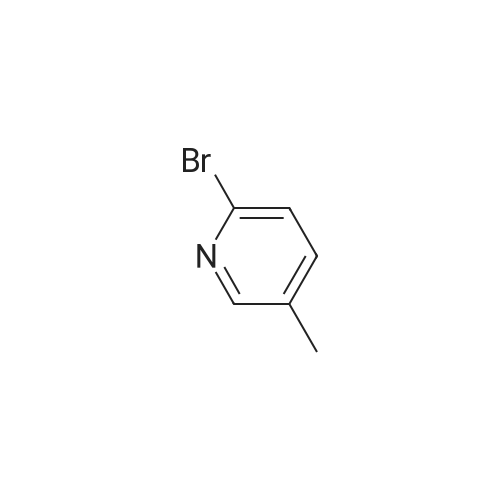
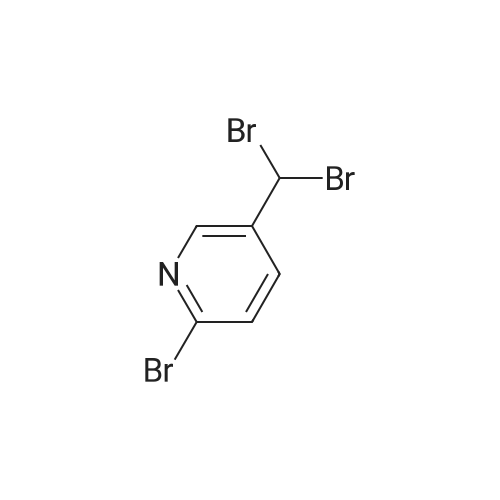
Synthesized from 2-bromo-5-methylpyridine (3.00 g, 17.40 mmol), NBS (3.41 g, 19.20 mmol) and DBPO (230 mg, 0.80 mmol) in carbon tetrachloride according to Method A. Yield: 2.56 g (59 %); lachrymatory yellow needles; 1 H NMR (CDCIs, 500 MHz): δΗ (ppm) = 4.14 (s, 2H), 7.47 (d, J = 8.2 Hz, 1 H), 7.59 (d, J = 8.2 Hz, 1 H), 8.38 (s, 1 H); MS (ESI): m/z = 252.37 [M+H]+.Methylpyridine was dissolved in 40 ml_ of dry carbon tetrachloride. To this solution was added /V-bromsuccinimide (NBS) (1.1 eq) and benzoyl peroxide (5 mol%) and the mixture was refluxed over night. After cooling, the succinimide was removed by filtration and the filtrate was concentrated under vacuum. The crude product was further purified by flash column chromatography on silica gel using a mixture of petroleum ether / ethyl acetate (95:5) as eluent. |
| 52.6% |
With N-Bromosuccinimide; dibenzoyl peroxide; In Carbon tetrachloride; at 80℃; for 2h;Inert atmosphere; |
To a solution of 169-51 (5.0 g, 29.1 mmol) in Cd4 (40 mL) was added NBS (5.22 g, 29.1 mmol) and BPO (350 mg, 1.4 mmol). The reaction mixture was stirred at 80 C under an atmosphere of nitrogen for 2 hours. The mixturewas diluted with DCM, washed with water and brine, dried, and concentrated to dryness. The remaining residue was purified by column chromatography on silica gel (eluted with PE/EtOAc = 120:1) to afford 169-S2 (3.81g, 52.6% yield) as a yellow oil. LC/MS (ESI) m/z: 250 (M+H)t |
| 50% |
With N-Bromosuccinimide; 2,2'-azobis(isobutyronitrile); In Carbon tetrachloride; at 75℃; for 2h;Inert atmosphere; |
Step 1) 2-bromo-5-(bromomethyl) pyridine To a solution of 2-bromo-5-methylpyridine (17 g, 100 mmol) in carbon tetrabromide (250 mL) was added AIBN (164 mg, 1mmol) and the mixture was heated to 75 C, and then NBS (26.7 g, 150 mmol) was added, and the mixture was stirred for 2 h. TLC showed that the reaction was complete, water (150 mL) was added and extracted with ethyl acetate (150 mL x 3). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated to give 3.0g of the residue, which was purified by column chromatography (silica gel 200-300 mesh, PE/EA= 1000:1 to 10:1) to give intermediate 2-bromo -5-(bromomethyl) pyridine (13 g, 50% yield). MS (ESI), m/z, 250.9 [M+1]+. |
| 49% |
With N-Bromosuccinimide;dibenzoyl peroxide; In Carbon tetrachloride; for 4h;Heating / reflux; |
2-Bromo-5-bromomethylpyridine. 2-Bromo-5-methylpyridine (5.00 g, 29.1 mmoles) and N-bromosuccinimide (5.22 g, 29.3 mmoles) were dissolved in carbon tetrachloride (40 mL) under nitrogen. Benzoyl peroxide (0.35 g, 1.4 mmoles) was added and the mixture heated at reflux for four hours. The mixture was cooled to room temperature, filtered, and washed with NaHCO3/H2O. The mixture was adsorbed onto silica gel and then chromatographed. eluting with a gradient of hexane to 10% ethyl acetate/hexane. Pure fractions were combined and concentrated to provide the desired mono-brominated product as a pale yellow solid, 3.60 g (49%). LC/MS (M+H)+ m/z=249.8, 251.8, 253.8. |
| 49% |
|
Step A2-Bromo-5-bromomethylpyridine. 2-Bromo-5-methylpyridine (5.00 g, 29.1 mmoles) and N-bromosuccinimide (5.22 g, 29.3 mmoles) were dissolved in carbon tetrachloride (40 mL) under nitrogen. Benzoyl peroxide (0.35 g, 1 .4 mmoles) was added and the mixture heated at reflux for four hours. The mixture was cooled to room temperature, filtered, and washed with NaHC03/H2O.The mixture was adsorbed onto silica gel and thenchromatographed. eluting with a gradient of hexane to 10% ethyl acetate/hexane. Pure fractions were combined and concentrated to provide the desired mono-brominated product as a pale yellow solid, 3.60 g (49%). LC/MS (M+H)+ m/z = 249.8, 251.8, 253.8. |
| 46% |
With N-Bromosuccinimide; 2,2'-azobis(isobutyronitrile); In dichloromethane; at 55℃; for 6h;Heating; Irradiation; |
Example 16 Step 1: A mixture of 2-bromo-5-methylpyridine (10 g, 58 MMOL), N-bromosuccinimide (15.5 g, 87.2 MMOL), and AZOBISISOBUTYRONITRILE (0.25 g) in anhydrous CH2CI2 (100 ml) was heated at 55 C under irradiation (200W lamp) for 6 h. The mixture was cooled down to RT, diluted with CH2CI2 (200 ML), washed with saturated NaHCO3 solution, dried (MGS04), filtered and concentrated. The residue was subjected to silica gel flash chromatography (5<7% EtOAc/hexanes) to afford the product (6.75 g, 46%). |
| 20% |
With N-Bromosuccinimide; dibenzoyl peroxide; In Carbon tetrachloride; for 19h;Reflux; |
Synthesis Example 18 N-[1-((6-bromopyridin-3-yl)methyl)pyridin-2(1H)-ylidene]-2,2,2-trifluoroacetamide (Compound 231) An amount of 500 mg (2.92 mmol) of 2-bromo-5-methylpyridine was dissolved in 15 mL of carbon tetrachloride, following which 623 mg (3.50 mmol) of N-bromosuccinimide and 10 mg of benzoyl peroxide were added and the mixture was refluxed under heating for 19 hours. Following reaction completion, the reaction mixture was returned to room temperature and concentrated under reduced pressure, then purified by silica gel column chromatography (hexane/ethyl acetate=19:1), giving 143 mg of 2-bromo-5-bromomethylpyridine (yield, 20%). 1H-NMR (CDCl3, δ, ppm): 4.42 (2H, s), 7.47 (1H, d), 7.59 (1H, dd), 8.38 (1H, d) |
| 20% |
With N-Bromosuccinimide; dibenzoyl peroxide; In Carbon tetrachloride; for 19h;Reflux; |
500 mg (2.92 mmol) of 2-bromo-5-methylpyridine was dissolved in 15 ml of carbon tetrachloride, 623 mg (3.50 mmol) of N-bromosuccinimide and 10 mg of benzoyl peroxide were added thereto, and the resulting mixture was heated and refluxed for 19 hours. After the reaction was completed, the reaction solution was returned to room temperature, concentrated under reduced pressure and purified by silica gel column chromatography (hexane: ethyl acetate = 19:1) to obtain 143 mg (yield 20%) of 2-bromo-5-bromomethylpyridine. [0250] 1H-NMR (CDCl3, δ, ppm): 4.42(2H, s), 7.47(1H, d), 7.59(1H, dd), 8.38(1H, d) |
| 20% |
With N-Bromosuccinimide; dibenzoyl peroxide; In Carbon tetrachloride; for 19h;Reflux; |
500 mg (2.92 mmol) of 2-bromo-5-methylpyridine was dissolved in 15 ml of carbon tetrachloride, 623 mg (3.50 mmol) of N-bromosuccinimide and 10 mg of benzoyl peroxide were added thereto, and the resulting mixture was heated and refluxed for 19 hours. After the reaction was completed, the reaction solution was returned to room temperature, concentrated under reduced pressure and purified by silica gel column chromatography (hexane: ethyl acetate = 19:1) to obtain 143 mg (yield 20%) of 2-bromo-5-bromomethylpyridine. [0240] 1H-NMR (CDCl3, δ, ppm) : 4.42(2H, s), 7.47 (1H, d), 7.59 (1H, dd), 8.38 (1H, d) |
| 20% |
With N-Bromosuccinimide; dibenzoyl peroxide; In Carbon tetrachloride; for 19h;Reflux; |
In 15 ml of carbon tetrachloride, 500 mg (2.92 mmol) of 2-bromo-5-methylpyridine was dissolved. To this solution, 623 mg (3.50 mmol) of N-bromosuccinimide and 10 mg of benzoyl peroxide were added, followed by heating under reflux for 19 hours. After completion of the reaction, the reaction liquid was returned to room temperature, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane:ethyl acetate=19:1). Thus, 143 mg of 2-bromo-5-bromomethylpyridine was obtained (Percentage Yield: 20%). 1H-NMR (CDCl3, δ, ppm): 4.42 (2H, s), 7.47 (1H, d), 7.59 (1H, dd), 8.38 (1H, d) |
|
With N-Bromosuccinimide; azobisisobutyronitrile; In Carbon tetrachloride; ethyl acetate; |
Step A 2-Bromo-5-bromomethyl-pyridine 2-Bromo-5-methylpyridine (4.01 g, 23.31 mmol), NBS (5.19 g, 29.14 mmol), and AIBN (0.19 g, 1.17 mmol) were dissolved in CCl4 (46.6 mL, 23.31 mmol). The reaction mixture was heated to 75 C. for 4 h. The residue was quenched with water and extracted from EtOAc. The organic layer was dried over anhydrous MgSO4 and concentrated in vacuo in vacuo. The crude mixture was purified by flash column chromatography on silica gel eluted with 7% EtOAc/hexane to give 2.65 g of the title compound. MS m/e 251.9 (M+). |
|
With N-Bromosuccinimide; In Carbon tetrachloride; hexane; ethyl acetate; |
Step D Preparation of 2-bromo-5-(bromomethyl)pyridine In a three neck flask equipped with a stir bar and a reflux condenser, a solution of 2-bromo-5-methylpyridine (40.0 g, 0.225 mol) and N-bromosuccinimide (45.0 g, 0.252 mol) in carbon tetrachloride (800 mL) was irradiated with a 300 watt lamp. After 3 h, the solution was cooled, and the solvent removed in vacuo. The residue was partitioned between ethyl acetate and water, and the organic phase washed with saturated sodium chloride solution and dried over magnesium sulfate. The crude product was purified by flash chromatography on silica gel using 7% ethyl acetate in hexane. The title product was obtained as a white solid. |
|
With N-Bromosuccinimide; 2,2'-azobis(isobutyronitrile); In Carbon tetrachloride; |
Step 4: Preparation of 2-bromo-5-bromomethylpyridine. A solution of 296.3 g (1.72 mol) of 2-bromo-5-picoline from step 3 in 6 L of carbon tetrachloride was treated with 306.5 g (1.72 mol) of N-bromosuccinimide (NBS) and 28.3 g (173 mmol) of azobisisobutyronitrile (AIBN). The reaction was stirred at reflux under nitrogen for 3 h, filtered, and concentrated in vacuo providing 476 g of crude 2-bromo-5-bromomethylpyridine as a brownish yellow solid (NMR indicates that this material is only 69% monobromomethyl product): NMR (CDCl3) δ 4.42 (s, 2H), 7.48 (d, J=9 Hz, 1H), 7.60 (dd, J=9 and 3 Hz, 1H), 8.37 (d, J=3 Hz, 1H). |
|
With N-Bromosuccinimide; dibenzoyl peroxide; In Carbon tetrachloride; |
Step 2 2-bromo-5-bromomethyl pyridine To a flask was charged 2-bromo-5-methylpyridine (34.9 mmol, 6.0 g), N-bromosuccinimide (38.4 mmol, 6.83 g), benzoyl peroxide (3.49 mmol, 0.85 g) and carbon tetrachloride (60 mL). This solution was refluxed for 6 hr, cooled to ambient temperature and purified on a silica gel column. Eluted with ethyl acetate: hexane (1:9) to provide the title compound. FAB MS: calc: 250.9 found: 251.9. 1 H-NMR (CDCl3): 4.4 ppm (s, 2H); 7.5 ppm (d, 1H); 7.6 ppm (d, 1H); 8.4 ppm (s, 1H). |
|
With N-Bromosuccinimide;2,2'-azobis(isobutyronitrile); In Carbon tetrachloride; at 75℃; for 5h; |
To a suspension of 2-bromo-5-methyl-pyridine 11 (5.0Og, 29 mmol) and NBS (5.162g, 29 mmol) in CCl4 (40 mL) is added AIBN (0.477g, 2.9 mmol). The reaction is stirred at 75 0C for 5 hours and filtered. The filter cake is washed with CCl4, and the filtrate is evaporated to give a light yellow residue. |
|
With N-Bromosuccinimide; 2,2'-azobis(isobutyronitrile); In Carbon tetrachloride; for 1.5h;Heating / reflux; |
a) (S)-tert-Buty{ 2-amino-3-(6-bromopyridin-3-yl)propanoatePrepared according to the procedures detailed in Patent WO2006/127948.To a slurry of 2-bromo-5-methylpyridine (10.29 g) and N-bromosuccinimide (5.32 g) in carbon tetrachloride (150 mL) was added AIBN (200 mg) and the reaction vessel was purged with nitrogen. The reaction mixture was heated, under reflux, for 1.5 h then allowed to cool <n="54"/>to room temperature. The reaction mixture was filtered and the filtrate was concentrated in vacuo to give 2-bromo-5-(bromomethyl)pyridine (4 g). |
|
With N-Bromosuccinimide; dibenzoyl peroxide; In Carbon tetrachloride;Reflux; |
To a solution of 2-Bromo-5-methylpyridine (3.1 g, 17.4 mmol) in carbon tetrachloride (40 mL, 400 mmol) was added benzoyl peroxide (230 mg, 950 μmol) and NBS (3.4 g, 19.2 mmol). The resulting mixture was heated at reflux overnight. The mixture was cooled at 0 C. and the NBS was removed by filtration. The filtrate was concentrated to yield 2-bromo-5-bromomethylpyridine (4.6 g), which was used directly in the next step. |
|
With N-Bromosuccinimide;dibenzoyl peroxide; In Carbon tetrachloride;Inert atmosphere; Reflux; |
To a solution of 2-bromo-5-methylpyridine ((3.1 g, 17.4 mmol, 1 eq.) in carbon tetrachloride (40 mL, 400 mmol) was added benzoyl peroxide (230 mg, 950 μmol) and NBS (3.4 g, 19.2 mmol, 1.1 eq.). The resulting mixture was heated at reflux overnight. The mixture was cooled at 0 C. and filtered. The filtrate was concentrated to yield 2-bromo-5-bromomethylpyridine (4.6 g), which was used without further purification. |
|
With N-Bromosuccinimide; [2-(1,1'-biphenyl)-4-yl]-5-phenyloxazole; In Carbon tetrachloride; at 80℃; for 16h;Inert atmosphere; |
To a solution of 2-bromo-5-methyl-pyridine (20 g, 1 16.96 mmol) in CCL, (200 mL) was added NBS (21 .56 g, 122.80 mmol) and BPO (0.4 g, 1 %) under N2. The mixture was stirred at 80 C for 16 hr, then cooled to r.t., filtered and concentrated to afford the desired crude compound which was used directly in the next step. LCMS: m/z 252 (M+l )+. |
|
With N-Bromosuccinimide; In 1,2-dichloro-ethane; at 85℃; for 0.5h; |
2-bromo-5-methyl-pyridine (70.0 mmol) and N-bromosuccinimide (80.0 mmol) were dissolved in 1,2- dichioroethane (150 ml) and to this mixture 2,2’-azobis(2- ethylpropionitrile) (1.50 mmol) was added. The reaction mixture was heated under reflux at 85 C. for 15 minutes and next portion of 2,2’-azobis(2-methylpropionitrile) (1.50 mmol) was added and reaction mixture was heated at 85 C. for further 15 minutes. After cooling to room temperature was the reaction mixture kept at 5 C. for 2 hours and the precipitate was filtered off and washed with small amount of 1 ,2-dichlo- roethane. The filtrate was evaporated under reduced pressure and the crude product was used for further reaction step without purification. The crude 2-bromo-5-bromomethyl-py- ridine was dissolved in chloroform (100 ml) and urotropine (70.0 mmol) was added. The reaction mixture was stirred at room temperature for 16 hours. The precipitate was filtered off, washed with small amount of chloroform and dried on air. The crude urotropine salt was refluxed in a mixture of conc. ammonium hydroxide (12 ml) and water (80 ml) for 90 minutes and afier cooling to room temperature, 40% formaldehyde (5.0 ml) was added with stirring. The precipitate was filtered off, washed with ice-cold water and dried in vacuum dessicator. The crude product was crystallized from ethanol. Yield: 40% m.p. 105-106 C. Elemental analysis: Calcd. for C5H7HrN2 (187.04): C, 38.53; H, 3.77; N, 14.98. Found: C, 38.22; H, 3.72; N, 14.71. HPLC-MS (ESI+): 188.02(97.2%). ‘H NMR (DMSO, d5): 4.04 (t, J=5.67, 2H, CH2), 7.71 (d, J=8.19, 1H,ArH), 7.95 (dd, J=8.19, J’=i.95, 1H,ArH), 8.51 (d, J=i .95, 1H, ArH), 8.74 (s(br), 2H, NH2). |
|
With N-Bromosuccinimide; 2,2'-azobis(isobutyronitrile); In acetonitrile; at 90℃; for 12h;Inert atmosphere; |
To a solution of 2-bromo-5-methyl-pyridine (20.0 g, 116 mmol) in acetonitrile (300 mL) was added 2,2-azobisisobutyronitrile (1.91 g, 11.63 mmol) and N-bromosuccinimide (24.8 g, 139 mmol), and the mixture was stirred at 90 C for 12 hrs under nitrogen atmosphere. On completion, the reaction was concentrated to give a residue. The residue was purified by chromatography on silica gel (petroleum ether: ethyl acetate = 10: 1 to 5: 1) to give the title compound. MR (400MHz, CDC13) δ = 8.40 (d, J = 2.4 Hz, 1H), 7.62 (dd, J = 2.6, 8.2 Hz, 1H), 7.51 (d, J= 8.3 Hz, 1H), 4.42 (s, 2H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
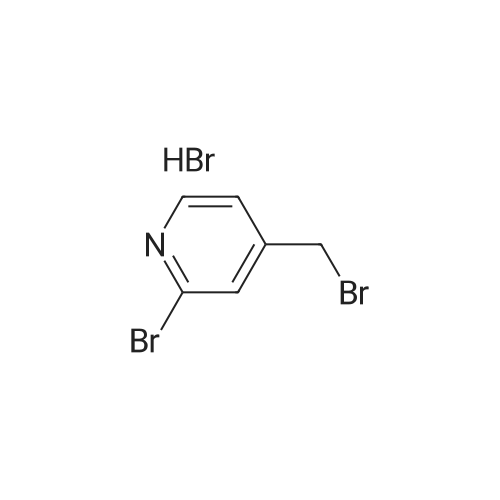
HazMat Fee +

 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping