845614-12-2
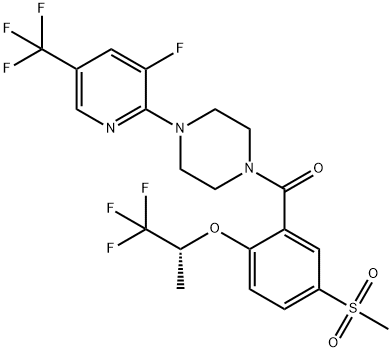 845614-12-2 結構式
845614-12-2 結構式
基本信息
[4-(3-氟-5-三氟甲基吡啶-2-基)哌嗪-1-基][5-甲基磺?;?2-[((R)-2,2,2-三氟-1-甲基乙基)氧基]苯基]甲酮
RG 1678 (R enantioMer)
RG-1678 (R enantioMer)
RO4917838 (R enantioMer)
Bitopertin (R enantioMer)
RG1678 (R ENANTIOMER)
RG 1678 (R ENANTIOMER)
RG-1678 (R ENANTIOMER)
RO4917838 (R ENANTIOMER)
[4-(3-Fluoro-5-trifluoromethylpyridin-2-yl)piperazin-1-yl][5-Methylsulfonyl-2-[((R)-2,2,2-trifluoro-1-methylethyl)oxy]phenyl]methanone
Methanone, [4-[3-fluoro-5-(trifluoromethyl)-2-pyridinyl]-1-piperazinyl][5-(methylsulfonyl)-2-[(1R)-2,2,2-trifluoro-1-methylethoxy]phenyl]-
物理化學性質
常見問題列表
IC50: 25 nM (GlyT1)
Bitopertin (RG1678) competitively blocks [ 3 H]ORG24598 binding sites at human GlyT1b in membranes from Chinese hamster ovary cells. Bitopertin potently inhibits [ 3 H]glycine uptake in cells stably expressing hGlyT1b and mGlyT1b, with IC 50 values of 25±2 nM and 22±5 nM, respectively (n=6). Conversely, Bitopertin has no effect on hGlyT2-mediated glycine uptake up to 30 μM concentration. Bitopertin has high affinity for the recombinant hGlyT1b transporter. Under equilibrium conditions (1 h at room temperature), Bitopertin displaces [ 3 H]ORG24598 binding with a K i of 8.1 nM. In hippocampal CA1 pyramidal cells, Bitopertin enhances NMDA-dependent long-term potentiation at 100 nM but not at 300 nM. Additional profiling revealed that Bitopertin (RG1678) has an excellent selectivity profile against the GlyT2 isoform (IC 50 >30 μM) and toward a panel of 86 targets including transmembrane and soluble receptors, enzymes, ion channels, and monoamine transporters (<41% inhibition at 10 μM is measured for all targets).
Bitopertin (RG1678) dose-dependently increases cerebrospinal fluid and striatal levels of glycine measured bymicrodialysis in rats. Additionally Bitopertin attenuates hyperlocomotion induced by the psychostimulant D-amphetamine or the NMDA receptor glycine site antagonist L-687,414 in mice. Bitopertin also prevents the hyper-response to D-amphetamine challenge in rats treated chronically with phencyclidine, an NMDA receptor open-channel blocker. Administration of vehicle has no effect on extracellular levels of striatal glycine, which remained constant throughout the experiment. In contrast, p.o. administration of Bitopertin (1-30 mg/kg) produced a dose-dependent increase in extracellular glycine levels. Bitopertin 30 mg/kg produces glycine levels 2.5 times higher than pretreatment levels. A similar dose-dependent increase in glycine concentration is observed in the CSF of rats treated p.o. with Bitopertin (1-10 mg/kg) compared with vehicle-treated animals, 3 h after drug administration. Interestingly, the level of CSF glycine increase 3 h after Bitopertin dosing is very similar to the increase in the microdialysis experiment at the same time point. In vivo pharmacokinetic studies in rat and monkey reveals that Bitopertin (RG1678) has, in both species, a low plasma clearance, an intermediate volume of distribution, a good oral bioavailability (78% for rat, 56% for monkey), and a favorable terminal half-life (5.8 h for rat, 6.4 h for monkey). The plasma protein binding is high in the two preclinical species (97%) and in human (98%). The CNS penetration of Bitopertin in rat (brain/plasma=0.7) is better than that in mouse (brain/plasma=0.5).